
 Sustainability has become a key trend in drug manufacturing. As we’ve previously discussed (here and here), this is being driven by environmental/good corporate stewardship efforts as well as various cost considerations. From greening supply chains to reductions in carbon emissions, pharmaceutical companies are increasingly taking steps to incorporate the principles of green chemistry into every aspect of their business.
Sustainability has become a key trend in drug manufacturing. As we’ve previously discussed (here and here), this is being driven by environmental/good corporate stewardship efforts as well as various cost considerations. From greening supply chains to reductions in carbon emissions, pharmaceutical companies are increasingly taking steps to incorporate the principles of green chemistry into every aspect of their business.
With organizational spotlights focused on sustainable practices, Environment, Health, and Safety (EHS) departments are receiving a great deal of attention.
The Challenging Role of an EHS Department
Although ecological responsibility and sustainability are popular buzzwords today, EHS teams don’t have an easy job. Much of pharma’s success, in fact, rests on the EHS department. From a ‘green business’ perspective, the department is responsible for developing and implementing policies that encourage sustainable practices throughout an organization. They also monitor the progress of those efforts.
From a ‘green chemistry’ perspective, they work closely with process and product chemists to ensure the safety of manufacturing processes. Without a competent team, the safety of the workplace, the employees, and the environment is at risk.
Effective EHS policies keep operations running smoothly toward the goal of sustainability, ultimately creating a healthy balance between risk mitigation and adherence to sustainable green principles.
EHS and Continuous Improvement
Even as EHS teams achieve successes and make strides in supporting sustainability, they remain aware that their work isn’t once-and done. Rather, it is an ongoing process focused on continuous improvement. Key functions of EHS team members include evaluating processes and practices, focusing on employee education through green training and setting measurable goals for green actions.
EHS Focus on Employee Health
To prioritize occupational health, an EHS team should offer training that covers personal hygiene, health monitoring, first aid knowledge, and adherence to COVID protocols. These health-related issues recognize the importance of human workers and are indispensable to the protection of company manpower. Many pharmaceutical companies are still rebounding from the detrimental effects of COVID-19 on the workforce. Protective measures that preserve employee health are crucial.
Additional EHS training that focuses on other areas of concern – such as chemical safety and emergency procedure – are also vital. The success of the safety-based employee education is often evidenced by a reduction in the number of negative incidents and failed audits that occur.
 Teams can monitor and facilitate company sustainability through:
Teams can monitor and facilitate company sustainability through:
Drug Manufacturing Sustainability Practices
The sustainability practices EHS teams employ are largely directed towards the reduction of waste output. Below are additional practices that EHS teams use to promote sustainability:
Despite the challenges associated with greener API manufacturing, some drug API manufacturers – including Neuland Labs – are leveraging better synthetic route design and sourcing alternate chemicals, reagents or precursors. The results can include less effluent and pollution, higher yields, shorter processing times, the use and storage of lower volumes of volatile chemicals, and the ability to downsize manufacturing infrastructure.
EHS, Sustainability & Neuland Labs
All of Neuland’s manufacturing units have adopted policies that protect the environment – including climate change and energy policies to reduce the company’s carbon footprint and Zero Waste Discharge policies to help minimize waste to landfills.
One key aspect of sustainable chemistry focuses on minimizing waste disposal via incineration. Working towards a goal of 0% incineration of waste, we send our spent solvent and other material waste to cement manufacturers, who use co-processing technology to convert this waste to an auxiliary fuel.
We achieved a recovery rate of 80-85% in FY 2021 using sophisticated solvent recovery systems. A wastewater treatment plant with higher capacity also came online, enabling the efficient management of additional load on the effluent system due to operational surges.
Some of the additional changes we’ve implemented are:
By assuming global good stewardship practices and incorporating EHS-focused actions, the pharmaceutical industry is well-positioned to optimize opportunities to protect the world’s citizens and ecological systems.
Related Blog Content
EHS Stewardship: Green Chemistry, Healthy People, Healthy Workplace
Green Generic Drug Substances: Increased Upfront Costs Offset by Long-Term Profitability
API Production: Building a Process Safety Culture
Resources
Learn more about Neuland’s Environmental, Health and Safety (EHS) Team.
Learn about Corporate Social Responsibility at Neuland Labs.
 Green chemistry principles first emerged in the 1990s, and the pharmaceutical industry – especially larger, global organizations – began steadily incorporating its principles in subsequent years.
Green chemistry principles first emerged in the 1990s, and the pharmaceutical industry – especially larger, global organizations – began steadily incorporating its principles in subsequent years.
Here are some recent examples of how Big Pharma has embraced the opportunity of green chemistry:
Green Chemistry: Needs to be Made the Norm, Not the Exception
Green chemistry is no longer the sole preserve of Big Pharma MNCs. With global pollution levels rising at an alarming rate and worst-case climate scenarios playing out around the world, significant efforts are underway across the industry.
Much more remains to be done, however. Adopting green chemistry principles in pharmaceutical process design and development unfortunately still remains the exception rather than the rule – something which must be changed in order to fully transform the industry.
Conceptually, the aim is waste and hazard prevention, but green chemistry is more than just an environmentally responsible approach to chemical product applications and processes. It has become critically important to the corporate bottom line, as well – hence its increasing adoption.
To minimize pharma’s adverse ecological impact and eliminate or reduce the global production and use of harmful chemical contaminants, pharmaceutical industry leaders are concentrating their efforts on embracing green chemistry concepts.
Pharmaceutical chemists are aware of the correlation between an increase in the organic syntheses of life-changing pharma products and surges in hazardous waste generation. Generally, the response has been a growing focus on greener alternatives to current processes.
Assuming Ecological Stewardship
For Neuland, assuming ecological stewardship means we remain aware that the application of green principles must progress from the lab bench to commercial scales. As we have voiced during various presentations in the past, the research of greening methods in the drug industry must fundamentally include actual process research rather than only the exploration of process design changes.
Successfully putting green chemistry into practice at scale, however, demands a rethink of existing practices. Chief amongst them is ensuring consideration is given to research across the various stages in the manufacturing process. Rather than highlighting only downstream process changes, we’ve found it effective to also prioritize research into front-end processes to identify pathways which promote process efficiency.
 Focusing on Green Concepts
Focusing on Green Concepts
Green chemistry includes 12 principles— all designed to facilitate safety and reduce waste. As Neuland embraces these greening concepts, we are directing particular focus toward areas that apply to active pharmaceutical ingredients, such as atom economy, zero liquid discharge and chemical safety.
 As the pharmaceutical industry strives for sustainable development, Neuland continues to research and develop safer processes for products, focusing on reducing waste generation, and improving efficiencies. Not only will these strategies lessen pharma’s ecological footprint, but they can also realize significant cost savings in the long run by decreasing the need for waste disposal and reagent procurement.
As the pharmaceutical industry strives for sustainable development, Neuland continues to research and develop safer processes for products, focusing on reducing waste generation, and improving efficiencies. Not only will these strategies lessen pharma’s ecological footprint, but they can also realize significant cost savings in the long run by decreasing the need for waste disposal and reagent procurement.
Related Blog Content
EHS Stewardship: Green Chemistry, Healthy People, Healthy Workplace
Green Generic Drug Substances: Increased Upfront Costs Offset by Long-Term Profitability
API Production: Building a Process Safety Culture
Resources
Learn more about Neuland’s Environmental, Health and Safety (EHS) Team.
Learn about Corporate Social Responsibility at Neuland Labs.
 As a follow-up to our recent two-part series on Regulatory Starting Materials Sourcing & Supplier Management, we sat down with Senior Vice President of Supply Chain Management at Neuland Labs Dr. Sundar Narsimhan to get his take on finding and qualifying suppliers for regulatory starting materials (RSMs).
As a follow-up to our recent two-part series on Regulatory Starting Materials Sourcing & Supplier Management, we sat down with Senior Vice President of Supply Chain Management at Neuland Labs Dr. Sundar Narsimhan to get his take on finding and qualifying suppliers for regulatory starting materials (RSMs).
What was Dr. Sundar’s key takeaway? Invest the time necessary in supply chain management of RSMs upfront to minimize the likelihood of problems downstream – and across the drug lifecycle.
![]() What tips and tricks can you share for identifying and qualifying a supplier of regulatory starting materials?
What tips and tricks can you share for identifying and qualifying a supplier of regulatory starting materials?
In my 15 years in the pharma industry, a sustained search of the supply base usually leads to the jackpot. Fine-tooth combing through high-quality databases like DWCP and Row2Technologies can unearth excellent sourcing options for any given API and its intermediates.
That being said, results can be hit or miss if NECs are involved, and commercial RSM options are limited. In that case, it may be up to the R&D team to develop a cost-effective manufacturing process for the starting material to facilitate commercial external manufacturing.
![]() In your opinion, how crucial is supply chain management when applied to regulatory starting materials?
In your opinion, how crucial is supply chain management when applied to regulatory starting materials?
The criticality of supply chain management in the procurement of RSMs can’t be overstated. Lives are literally at stake, so the first objective of supply chain management is to ensure consistent application of cGMP practices.
As a professional in the pharmaceutical industry, the industry’s objectives of preventing deaths and illnesses while improving health have always been a point of pride, and I believe effective supply chain management is a huge part of that.
![]() What’s the most important step in sourcing and supplier management?
What’s the most important step in sourcing and supplier management?
Due diligence has always been an essential part of RSM procurement. If you’re looking to establish a long-term relationship with a supplier, you can’t afford to skimp on due diligence. Do due diligence on your due diligence, and you should be good to go with RSMs.
![]() How do contract manufacturers like Neuland align their sourcing strategy with clients’ requirements?
How do contract manufacturers like Neuland align their sourcing strategy with clients’ requirements?

Flexibility and proactiveness are the biggest secret ingredients. For instance, at Neuland, our global sourcing strategy means we have a wider reach when it comes to ensuring the desired regulatory status and cost competitiveness for each client. Our regulatory affairs and other teams have a wealth of experience with RSM sourcing, which provides greater confidence to clients seeking to mitigate business risks.
![]() Any parting words of wisdom to pharmaceutical companies regarding supply chain management of RSMs?
Any parting words of wisdom to pharmaceutical companies regarding supply chain management of RSMs?
Don’t overlook this critical upstream process because it can potentially impact downstream processes. That’s a key reason why you should always work with an API contract manufacturer that is familiar with RSMs, has well-established sourcing procedures and understands the chemistry. The benefits include better traceability from starting material to final drug substance, reduced time to commercial launch, lower development costs, improved supply chain security, and more.
Learn more about regulatory starting materials. For questions on regulatory starting materials and supply chain management, contact the Neuland team.
In Part I of this two-part series, we explored sourcing and supplier management of Regulatory Starting Materials (RSMs), and how effective designation and justification of RSMs begins with supplier market analysis, feasibility studies, lab validation, and source differentiation.
These stages reduce the available supplier options. But there are additional steps to take on the path to a commercial launch. This brings us to Part II of our in-depth dissection of the selection process for RSMs.
The initial stages of sourcing and supply management of RSMs generate massive amounts of data. Therefore, API manufacturers have a significant amount of information at their disposal for shortlisting of sources. Due diligence then kicks in to ensure only verified data is used when further narrowing the shortlist. Performing due diligence is vital for critical starting materials to ensure accurate risk assessment.
The first step in data verification involves getting information directly from the supplier, in addition to information sourced from external sources. A tailored and detailed supplier questionnaire is typically sent out, and the collected information should match details from external sources.
The second step in this process involves a site visit by the due diligence team. This allows for more detailed verification of critical aspects, including capacities and infrastructure, health, safety, quality culture, and general compliance.
What the due diligence team finds on the ground should corroborate previously collected information. The assurance of contracting with the right supplier can only happen by conducting appropriate due diligence beforehand. The combined data is then condensed into a report. Ultimately, the final go or no-go decision rests with senior management.
At this stage, teams tasked with the selection of RSMs turn to audit plan implementation and plant validation. Audit plans and validation are necessary to support the final ‘Go’ or ‘No-Go’ decision. Both processes should be tailored to the particular chemical, precursor, or intermediate incorporated into the API manufacturing process.
Generally, an audit is based on key parameters such as material classification, risk level as identified during due diligence, assurance of supply at a commercial scale, and the acceptance criteria for regulatory approval.
 The audit plan is a valuable quality assessment tool that ensures the procurement process is headed in the right direction in the final stages. It culminates in an audit report that will potentially drive the finalization of the quality assessment and signing of the quality agreement.
The audit plan is a valuable quality assessment tool that ensures the procurement process is headed in the right direction in the final stages. It culminates in an audit report that will potentially drive the finalization of the quality assessment and signing of the quality agreement.
With plant validation, the central focus is procuring starting materials for the commercial phase. The validation process encompasses the production of 3-4 batches of the API to ensure process repeatability and establish if the yield, quality, and impurity profiles generated from prior lab experiments remain acceptable at the commercial scale.
Plant validation also includes the validation of equipment, labs, and processes as part of regulatory compliance for pharmaceutical manufacturing facilities. Doing this early can reduce time-to-market and increase budget optimization.
Data is consistently generated at each stage of the procurement process. This presents an opportunity for continuous data analysis to better ensure compliance with regulatory standards. In other words, a systematic evaluation of collected data allows for the judicious selection of RSMs and increases the chances of regulatory acceptance.
For instance, if a synthetic route has been developed using the proposed RSM, data analysis shows if there are enough chemical steps from the final drug substance. Shorter synthetic routes are unpopular with regulators since they typically don’t leave enough time or space to effectively remove impurities and isolate APIs.
 Once an analysis of the data shows the appropriate level of alignment required for commercial approval, it’s time for regulatory filing and review. Regulatory review of the proposed RSM generally depends on the location and the type of regulatory agency involved. So, knowing location-specific regulations and regulator protocols helps with accurate data analysis and anticipation of potential risks.
Once an analysis of the data shows the appropriate level of alignment required for commercial approval, it’s time for regulatory filing and review. Regulatory review of the proposed RSM generally depends on the location and the type of regulatory agency involved. So, knowing location-specific regulations and regulator protocols helps with accurate data analysis and anticipation of potential risks.
Data analysis and regulatory filing are better off performed before developing downstream processes, since failures in upstream processes could cause an expensive redo of downstream development work.
The sourcing and supply chain management of regulatory starting materials is a critical factor in the balancing act of meeting or exceeding regulatory requirements – while maximizing commercialization and de-risking supply. Investments made into the process allow stakeholders needs to be addressed and operational sustainability to be met.
 Supply chain management is an essential chapter in the pharmaceutical manufacturing handbook. Managing regulatory starting materials (RSMs) seeks to maximize commercialization and sustainability while meeting and/or exceeding regulatory requirements.
Supply chain management is an essential chapter in the pharmaceutical manufacturing handbook. Managing regulatory starting materials (RSMs) seeks to maximize commercialization and sustainability while meeting and/or exceeding regulatory requirements.
Drugmakers must also ensure the final product conforms to the non-negotiable, pharmaceutical standards of excellence relating to quality, safety, and efficacy.
Admittedly, it’s a fine line between advancing the priorities of regulatory authorities and the interests of stakeholders.
This two-part blog series offers an exploration of the critical stages of sourcing and supplier management of regulatory starting materials.
Supply market analysis sets the stage for identifying and qualifying a supplier of RSMs. It develops an understanding of the essential factors of the market, and the information helps formulate the right 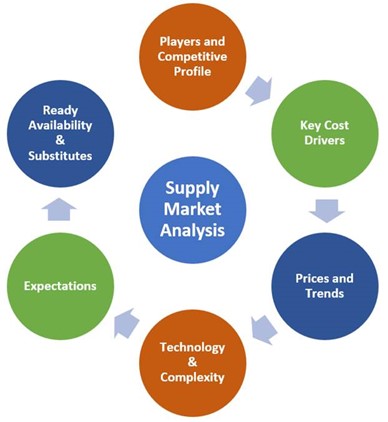 sourcing strategy for future, large-scale procurement initiatives.
sourcing strategy for future, large-scale procurement initiatives.
Because of this, supply market analysis should be considered obligatory before building new collaborations, especially if there’s a high degree of risk and/or value chain competitiveness. Generally, the rewards outweigh the resource investment for this analysis. Supply market analysis involves the following steps:
Basic manufacturer requirements of the soon-to-be-procured material should already be in place to ensure a narrower focus and efficient data gathering. The requisite details include product and material specifications, quantity, and regulatory classifications.
Supply market analysis should result in the beginnings of a viable supplier list. This, in turn, is a solid starting point for selecting suppliers and starting materials. Before manufacturing a new product, feasibility studies and lab validation are paramount.
 Feasibility studies and laboratory validation are used to assess whether starting materials are suitable. Feasibility studies shed light on the costs, routes, purity levels, process criticalities and technology deployment for the possible routes.
Feasibility studies and laboratory validation are used to assess whether starting materials are suitable. Feasibility studies shed light on the costs, routes, purity levels, process criticalities and technology deployment for the possible routes.
Suitable samples must be procured, assessed, and used to define the preferred route of synthesis. These steps define the number of critical process stages, the ease or difficulty of impurity purging, and the overall risk from start to finish.
After performing multi-batch feasibility studies and finalizing experimental work and production trials, a comprehensive validation report is compiled and signed off on. The next step is selecting the right RSM for procurement.
There’s a crucial connectivity between selecting suitable regulatory starting materials, and feasibility studies and laboratory validation. Remember, the validation stage involves the analytical testing of procured samples ahead of commercial production.
This stage of starting material designation and justification should arrive at a finalized shortlist of at least three suppliers. Generally, source selection uses wide-ranging acceptance criteria that cover the following:
These are some of the basics of supply management. In Part II, we’ll cover the later stages of sourcing and supply management with regard to regulatory materials.
 In 1905, George Santayana published his famous aphorism: “Those who cannot remember the past are condemned to repeat it.” More than 100 years later, this is could be an unofficial guiding principle of analytical drug chemistry. The entire point of analytical chemistry is to build on those discoveries which have come before.
In 1905, George Santayana published his famous aphorism: “Those who cannot remember the past are condemned to repeat it.” More than 100 years later, this is could be an unofficial guiding principle of analytical drug chemistry. The entire point of analytical chemistry is to build on those discoveries which have come before.
Genotoxic impurities? We’ve established thresholds and control strategies. Nitrosamines? Drugmakers are now mindful of their presence.
In the 100+ years since Santayana’s saying, our scientific knowledge of impurities has changed as radically and as quickly as the instrumentation and technology used to study them.
But the subtext here is also strikingly clear: with each passing discovery, we gain a little bit more insight into how much we still don’t know.
What we do know guides modern pharmaceutical science. We know impurities can appear in drug products at all stages of manufacturing, and we know that they can impact the safety or efficacy of a compound.
We also understand the primary sources of concern for impurities:

[For a full exploration of each of these categories – and many more – read PharmTech’s excellent 3-part rundown on impurities. While a bit dated (2012), it’s an excellent overview.]
As the role played by analytical chemistry in drug development has grown, our working body of knowledge – impurity formation, genotoxins, route design and much more – has allowed us to proactively confront impurities in an effort to ensure drug safety.
At the same time, it’s recognized that it is practically impossible to completely remove impurities during manufacturing. For that reason, manufacturers must leverage a variety of different strategies for effective impurity detection, quantification and control. Comprehensive impurity profiles should be developed to understand:
In a 2020 article in the International Journal of Environmental research and Public Health (Chemical Impurities: An Epistemological Riddle with Serious Side Effects), the authors wrote: “Although impurities are considered a nuisance in chemical synthesis, they are generally of little concern as long as their identity is clear and their amounts are under control.”
Xpurities: Exploring Unknown Impurities
Impurity control is clearly essential, but what about the impurities we don’t yet know about – the ones yet to be identified and characterized? They are often referred to as unidentified impurities “that can be identified only with qualitative analytical values (e.g., peak area, retention time, etc.), for which structural information is not yet available.”
The authors of the article labelled these “Xpurities” to distinguish them from known, identified impurities. They were reported to be “surprisingly common and constitutes a major issue in pharmaceutical research and practice.”
A PharmTech article last Fall on unknown impurities discussed the various technologies currently in-use for impurity detection, as well as what’s on the radar. Current methods for small molecule impurities, based on an orthogonal approach, tend to rely on chromatography with UV-detection. Mass spectrometry (MS) is also a popular approach, but older mass specs (e.g., single-quadrupole MS) face challenges due to lower resolution.
Other methods to isolate and characterize impurities in pharmaceuticals, as described in the Journal of Advanced Pharmaceutical Technology & Research include:
“Capillary electrophoresis, electron paramagnetic resonance, gas–liquid chromatography, HPLC, solid-phase extraction, liquid–liquid extraction, UV spectrometry, infrared spectroscopy, supercritical fluid extraction, NMR and RAMAN spectroscopy.
Among all hyphenated techniques, the most exploited techniques for impurity profiling of drugs are LC-MS, LC-NMR, LC-NMR-MS, GC-MS, and LC-MS.”
The next generation gas chromatography-MS (GC-MS) and high-resolution MS (HRMS) are being developed to work in tandem with other technologies to detect and identify impurities in samples.
An article on USP’s Quality Matters blog discussed nitrosamine contaminants in drug products and how the initial 2018 valsartan problem grew in scope and continues to impact supply chains today.
“The complexity and global nature of the pharmaceutical supply chain demands greater diligence, transparency, and collaboration between manufacturers and regulatory agencies around the world to protect patient safety. Collaborative efforts between these and other stakeholders, along with effective tools to detect and control impurity levels, will help safeguard patients’ continued access to safe medicines that deliver the intended therapeutic benefit.”
 Impurity detection and control is a dynamic practice, and it relies on process chemistry expertise, a best practices approach, and familiarity with the latest instrumentation, methods & capabilities.
Impurity detection and control is a dynamic practice, and it relies on process chemistry expertise, a best practices approach, and familiarity with the latest instrumentation, methods & capabilities.
The valsartan nitrosamine issue was a clearcut example of how we – as an industry – build upon new discoveries and understandings. Nitrosamines have been around for quite some time, but it was only more recently that it became an issue for the drug industry and regulators.
Check out our in-depth explainer on How the Valsartan Contamination Happened: Its Context & Implications, or our follow-up post tracking the regulatory changes in response to nitrosamine contaminants.
 We’ve arrived at a time in which real-time remote GMP inspections of drug manufacturing facilities are an actual thing – and not just an “it would be nice to have that” item on regulator wishlists.
We’ve arrived at a time in which real-time remote GMP inspections of drug manufacturing facilities are an actual thing – and not just an “it would be nice to have that” item on regulator wishlists.
Due to the global travel restrictions and the outbreak of COVID, all major regulatory agencies and health authorities are either considering – or are already performing – real-time remote review of manufacturing operations, equipment, facilities and relevant documentation (such as records and logbooks).
GMP Inspection Backlogs
The principle underlying the move towards remote inspection is that it will allow a better understanding of GMP compliance compared to existing remote procedures which look exclusively at documentation. But there are also capacity and backlog issues plaguing regulators – and notably the FDA, who had been struggling with the volume of foreign inspections pre-pandemic.
In December, Fierce Pharma (2022 forecast: Can the FDA whittle down its manufacturing site inspection backlog next year?) explored the FDA’s blend of digital, remote and in-person capabilities.
“Like many organizations during the pandemic, the FDA leveraged a mix of digital and remote tools. But while remote inspection tools are a vital resource during the pandemic, in-person inspections are ‘key’ to what the agency does, Denigan-Macauley added. Ultimately, the FDA needs to use those alternative oversight tools—like remote inspections, requesting records and relying on certain other regulators abroad—to supplement, rather than replace its traditional inspections.”
Remote GMP Inspections
Remote inspections are here, and major regulatory agencies around the globe are starting to gain experience alongside manufacturers.
Neuland has received a request from the EDQM to configure remote inspection for our Unit-2 facility, and discussions are at the early stages. Since so few pharma companies have had remote facility inspections during the pandemic, it’s still a novel experience.
 Most of us probably have an image in our minds of an employee walking around with an iPad or laptop, pointing things out to an online inspector.
Most of us probably have an image in our minds of an employee walking around with an iPad or laptop, pointing things out to an online inspector.
However, it isn’t that simple.
Thorough Preparation Pre-Inspection
Regulators have reported that remote inspections conducted in real-time demand better, more thorough preparation than typical on-site inspections. For example, it’s necessary to hold preparatory teleconferences with the FDA, followed by connectivity tests (“dry runs”) to ensure that all areas to be inspected are equipped with the necessary networks or infrastructure necessary to create a smooth, seamless process.
Circumstances will dictate the technical solutions needed for specific facility locations. Regulatory agencies have found that successful participating companies set up a combination of Wi-Fi networks, cellular networks or hybrid networks – and often a combination of the elements of each.
Regulators further found that a data-transmission rate over 100 kilobytes/second was capable of providing sufficiently steady video transmission – which was usually the case when using broadband LAN/WLAN in meeting rooms. Maintaining a consistent stream sometimes became a challenge when using wireless or cellular networks in manufacturing or more remote areas.
 For communication, sharing live video footage and reviewing documents, some agencies have begun using secure web conferencing applications, which limits security issues. Agencies report using both a primary and a secondary web conferencing application during inspections, both as part of a redundancy strategy and to allow inspectors to work in parallel. Both web conferencing tools could be installed on mobile devices with web cameras, which could then be used during the inspection.
For communication, sharing live video footage and reviewing documents, some agencies have begun using secure web conferencing applications, which limits security issues. Agencies report using both a primary and a secondary web conferencing application during inspections, both as part of a redundancy strategy and to allow inspectors to work in parallel. Both web conferencing tools could be installed on mobile devices with web cameras, which could then be used during the inspection.
Virtual Inspections – Virtually the Same?
All inspections make teams apprehensive. That’s natural, and our inspection of Unit-2 will be no different in most regards from any of the inspections we’ve hosted over last few decades – whether from FDA, EDQM, EMA, PMDA, WHO GMP, TGA or another agency.
 On another level, however, it is different. This is new technology – and a new way of working alongside one another – and everyone is still working out the knots. It’s exciting for Neuland to be part of this emerging process.
On another level, however, it is different. This is new technology – and a new way of working alongside one another – and everyone is still working out the knots. It’s exciting for Neuland to be part of this emerging process.
Outsourced Pharma had an excellent guest column earlier this year on preparing for a remote inspection, covering things to prepare in advance of the inspection, additional inspection planning tasks that need to occur and much more. (While we don’t yet have the experience of having undergone a virtual inspection, the information shared in the article seems to include many best practices and common inspection stumbling blocks.)
 A recent article at DCAT Value Chain Insights (Generics: What is the Next Industry Move?) shone a light on recent movements in the generics space by the MNCs, including Novartis and Pfizer.
A recent article at DCAT Value Chain Insights (Generics: What is the Next Industry Move?) shone a light on recent movements in the generics space by the MNCs, including Novartis and Pfizer.
The takeaway? It reads like a rollercoaster ride! Some are divesting, others are acquiring, everyone is restructuring and…well…generics just keep on growing their sales footprint.
Generic Restraint.
Generic medicines aren’t without their challenges. The two biggest issues restraining growth include:
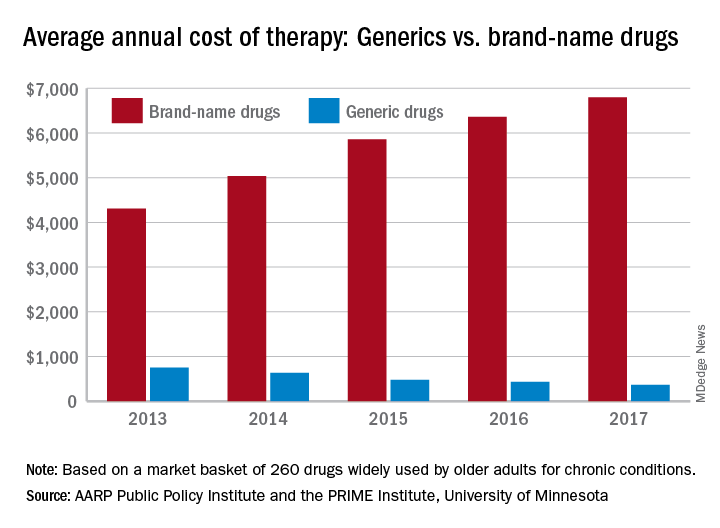 An article at Managed Healthcare Executive explored generic price tags, and shares “For many generics critical to patient care, there are no more savings to squeeze out…If we continue to do that, they will go away. Some manufacturers already produce them at a loss.”
An article at Managed Healthcare Executive explored generic price tags, and shares “For many generics critical to patient care, there are no more savings to squeeze out…If we continue to do that, they will go away. Some manufacturers already produce them at a loss.”
The chart (right) from MDEdge demonstrates the continued pricing pressures on generics even as brand-name drugs continue to rise.
For manufacturers, there are also supply chain dynamics at play impacting unit costs for those same products already facing pressure.
Shortages, massive cost spikes, and other weaknesses in the global supply chain – notably for precursors, intermediates and APIs – have become more visible over the last 2 years, and efforts to strengthen drug supply chains have been launched globally.
In a post earlier this year on generics penetration in global markets, we shared “[T]he attitude of physicians and pharmacists towards generics still varies a lot worldwide, even between Northern and Southern European countries. In the Nordic countries trust in the quality of generics is higher than that in the Southern countries, with concerns only over their taste, packaging and appearance, since this may affect patients’ understanding and acceptance.”
Generics: Consistent Growth Trajectory

Despite these challenges and the turbulence of M&A, generics are a study in constancy. They continue to post strong annual growth, both in terms of market size as a segment but also as a growing proportion of overall prescribed drugs. Global Generic Drugs Market (2020) highlights that generic drugs saw a worldwide compound annual growth rate (CAGR) of 8.7% between 2016 and 2020. This growth is projected to continue in the foreseeable future.
According to a Prescient & Strategic Intelligence market report published earlier this year: “The major factors responsible for the growth of the market include rising geriatric population, increasing number of patent expiration of branded drugs, surging cases of chronic and acute diseases, and growing R&D expenditure of biotech and pharma companies.”
 What Are Some of the Top Near-Term Drivers of the Generic Drug Market?
What Are Some of the Top Near-Term Drivers of the Generic Drug Market?
Generics are primed into the future to continue their ‘push pull’-driven growth. Regulatory oversight has – of course – been on the rise since the industry first embraced globalization decades ago. It has been on a sloped trajectory, and there is no reason to believe this trend will change. On the other hand, there have been regulatory moves which have strengthened the generics sector. Prioritization of generic drug applications is just one of the latest trends, stretching back to Hatch-Waxman and the launch of the space by regulators.
Likewise, pricing pressures are assumed to be here to stay, though in some cases the bottom may have been reached with certain products or therapeutic segments. And opportunities will lend some balance – whether from new IP or exclusivity period opportunities.
 Green initiatives may be slowly gaining some traction across the pharmaceutical industry, but there is still a long way to go. These are some astonishing insights from sciencehistory.org.
Green initiatives may be slowly gaining some traction across the pharmaceutical industry, but there is still a long way to go. These are some astonishing insights from sciencehistory.org.
Unfortunately, the history of green transformation across the drug lifecycle is spotty, at best.
Green Chemistry for Generics: Route Selection
Consider the lifecycle of most APIs on the market – from the point where a pre-clinical development candidate for an NCE first emerges up to the point much later in the lifecycle where a generic version gets the green light.
 The transition to a generic version of a drug has never become the recognized point at which processes are redesigned to consider greener synthetic routes. Many companies take the path of least resistance – altering processes just enough to satisfy the intellectual property team. Typically, these ‘knock-off’ routes are tweaked to yield some improvements in scalability or cost, since the original chemistry route was never optimized to be less wasteful or less environmentally impactful. But they aren’t ‘green processes’ by any measure, though they give the appearance of ‘improvement.’
The transition to a generic version of a drug has never become the recognized point at which processes are redesigned to consider greener synthetic routes. Many companies take the path of least resistance – altering processes just enough to satisfy the intellectual property team. Typically, these ‘knock-off’ routes are tweaked to yield some improvements in scalability or cost, since the original chemistry route was never optimized to be less wasteful or less environmentally impactful. But they aren’t ‘green processes’ by any measure, though they give the appearance of ‘improvement.’
The most effective processes involve green chemistry by design (GCbD). GCbD involves incorporating green chemistry principles earlier rather than later in the manufacture of generic drugs.
A well-conceived, second-generation green route is the secret ingredient behind sustainable and cost-effective commercial manufacture of generic drugs. Many manufacturers and drugmakers still consider ‘green chemistry’ an expense, rather than an investment. In reality, savvy manufacturers are passionate about developing greener synthetic routes during the early stages of an API’s life cycle.
Why?
Three reasons. Lower costs, better EHS outcomes, and shorter lead times.
Careful route selection at the process research stage – and not later during process development – is essential to the development of an appropriate process which addresses both waste generation minimization and value-add objectives.
Green Chemistry By Design Is Good For Business

No business is immune from the drive towards profitability. So, while the virtues of GCbD are praiseworthy from a stewardship perspective, profitability is always a primary driver – and green chemistry does come with an upfront price tag. This has yielded a reluctance among cost-conscious drugmakers to pursue green chemistry earlier in generics development.
Upfront cost concerns, however, obscure the reality of green chemistry outcomes, which generate long-term economic and environmental benefits which more than offset the high initial investment.
How so?
Let’s summarize the benefits associated with choosing green chemistry by design for generic APIs:
These benefits are possible largely due to new technologies and approaches used in process research. Here’s a peek at the technology used to develop greener processes for generic APIs.
For a more in-depth look at green chemistry by design, refer to this publication authored and presented to the scientific community by a Neuland team member. In addition, you can check out this presentation at the 2014 International Green Chemistry conference by a member of the Neuland Labs team.
 Neuland Labs and Green Chemistry
Neuland Labs and Green Chemistry
Why not design a greener, less wasteful synthetic route in the first place?
At Neuland, we believe green chemistry is a worthwhile endeavor for our clients, since careful design of API manufacturing processes goes hand in hand with cost-effectiveness. Our GCbD initiatives for Generic Drug Substances encompass benign by design chemistry, atom economy, and the use of specific enzymes and chiral substrates to boost yields.
As technological advances continue to leave their footprint on API manufacturing, we look forward to discovering more opportunities to develop greener and greener synthetic routes.
 Drug development is exciting. There’s no getting around it: being a part of an industry which aims to improve human health is a great motivator for waking up and heading to work.
Drug development is exciting. There’s no getting around it: being a part of an industry which aims to improve human health is a great motivator for waking up and heading to work.
And for those of us lucky enough to work for a contract manufacturer focused on a diverse range of segments, that feeling of satisfaction is often multiplied when we see the results of what we do having an impact around the world.
For this post, we’re sharing graphical representations of the therapeutic areas in which Neuland participates. Our APIs cross many therapeutic segments – from CNS and cardiovascular compounds to hematology, respiratory, rheumatology and more.
For our most recent list of APIs, along with a breakdown by stage of development (commercial, validation, development or evaluation) and regulatory status (US DMF, CEP/COS or DMF filed), download our latest Product List (pdf). Note that some of these products are still under patent protection in countries around the world and will only be available for supply after patient expiry.
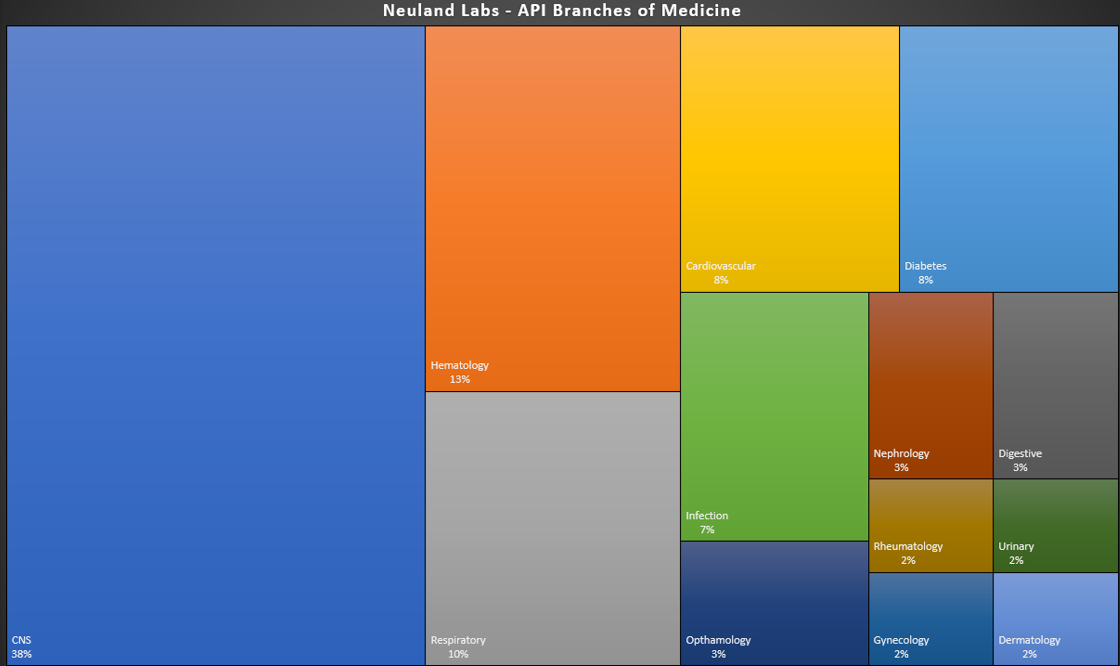 CNS Drug Targets
CNS Drug Targets
One noticeable attribute of the graphic above is our ostensible focus on CNS drugs. While we do have a substantial number of CNS compounds among our products, any characterization of our work as CNS-related would be a bit misleading. There are two reasons for this:
1. Neuland actually operates across an incredible diversity of segments, as seen in the graphic above.
2. Central Nervous System (CNS) therapeutics as a category is exceptionally broad, stretching from analgesics, anesthetics, anti-epileptics and anti-psychotics to drugs for Parkinson’s, MS, cancer, trauma, neurovascular diseases and much more. Within the ‘category’ of CNS, Neuland manufactures APIs for Alzheimer’s, Parkinson’s and migraine as well as antipsychotics, anticholinergics, anticonvulsants, anesthetics, etc. Each of these slivers of the CNS space is practically a market segment unto itself.

That being said, it is important to note that CNS is a huge opportunity. It also happens to be an exceptionally demanding segment for drugmakers. To date, science has gained only a partial understanding of certain CNS conditions, and still must grapple with overcoming the blood-brain barrier – a key hurdle to treatment.
From Advances in Drug Discovery for Central Nervous System Diseases (December 2020):
“Compared to other areas of drug discovery, the clinical failure rate for new drugs targeting the central nervous system (CNS) diseases is even higher. A study from the Tufts Center for the Study of Drug Development found that the success rate for CNS drugs, defined as final marketing approval by the FDA, was less than half the approval rate for non-CNS drugs (6.2 percent versus 13.3 percent) from 1995–2007. Additionally, the mean development time was greater, the time to approval following application submission for marketing approval was longer, and the number of CNS drugs given priority consideration by the FDA was significantly lower relative to non-CNS drugs.”
In 2020, the already-demanding characteristics of CNS development ran headlong into COVID. From A view into the central nervous system disorders market, at nature.com:
“Global sales of prescription and over-the-counter (OTC) central nervous system (CNS) disease-related products totaled $86 billion in 2019. Sales were predicted to grow in 2020, but the field has been one of those hardest hit by the COVID-19 pandemic. The total 2020 forecast fell by $1.4 billion between March and June of 2020, as social distancing and lockdown measures made clinics more difficult to access. But, despite the uncertainties caused by the pandemic, analysts predict that the CNS product market will expand to $101 billion in 2022 and to $131 billion in 2025.”
While drugs to treat CNS disorders face challenges, it is also a segment brimming with tremendous untapped potential. As we continue to improve our understanding of the biology of CNS conditions, novel therapeutic opportunities will emerge.








