How Pharmaceutical CDMO Supports DMF, ANDA & NDA Filing
Getting a drug approved takes more than strong clinical data because behind every successful filing sits detailed manufacturing documentation. That's where a pharmaceutical CDMO becomes essential.
From preparing Drug Master Files to compiling CMC sections for NDA submissions, CDMOs handle the regulatory groundwork that drug sponsors often lack in-house. In fact, 73% of FDA-approved drugs in 2025 outsourced their API manufacturing to contract partners.
This article explains how a pharmaceutical CDMO supports DMF, ANDA, and NDA filings — and why that support matters at every stage of the drug development regulatory process.
What Role Does a Pharmaceutical CDMO Play in Drug Regulatory Filings?
A pharmaceutical CDMO develops and manufactures active pharmaceutical ingredients for drug companies. But its role goes well beyond production.
CDMOs prepare regulatory documentation. They maintain GMP-compliant facilities. And they file critical dossiers with agencies like the FDA. Whether a company is submitting a DMF for an API, an ANDA for a generic drug, or an NDA for a novel therapy, the CDMO provides the manufacturing evidence that regulators require.
This includes process validation data, impurity profiles, stability studies, and analytical method reports.
For biotech firms without in-house manufacturing, this support is even more critical. Most emerging biopharma companies outsource drug development entirely. Building a cGMP facility from scratch costs $200 million or more. That's capital better spent on R&D and clinical programs.
Example: A mid-stage biotech developing a peptide-based oncology drug may have strong Phase 2 data but no manufacturing infrastructure. A pharmaceutical CDMO steps in to produce clinical-grade API, compile the drug substance dossier, and prepare the facility for FDA inspection — all before the NDA is even submitted.
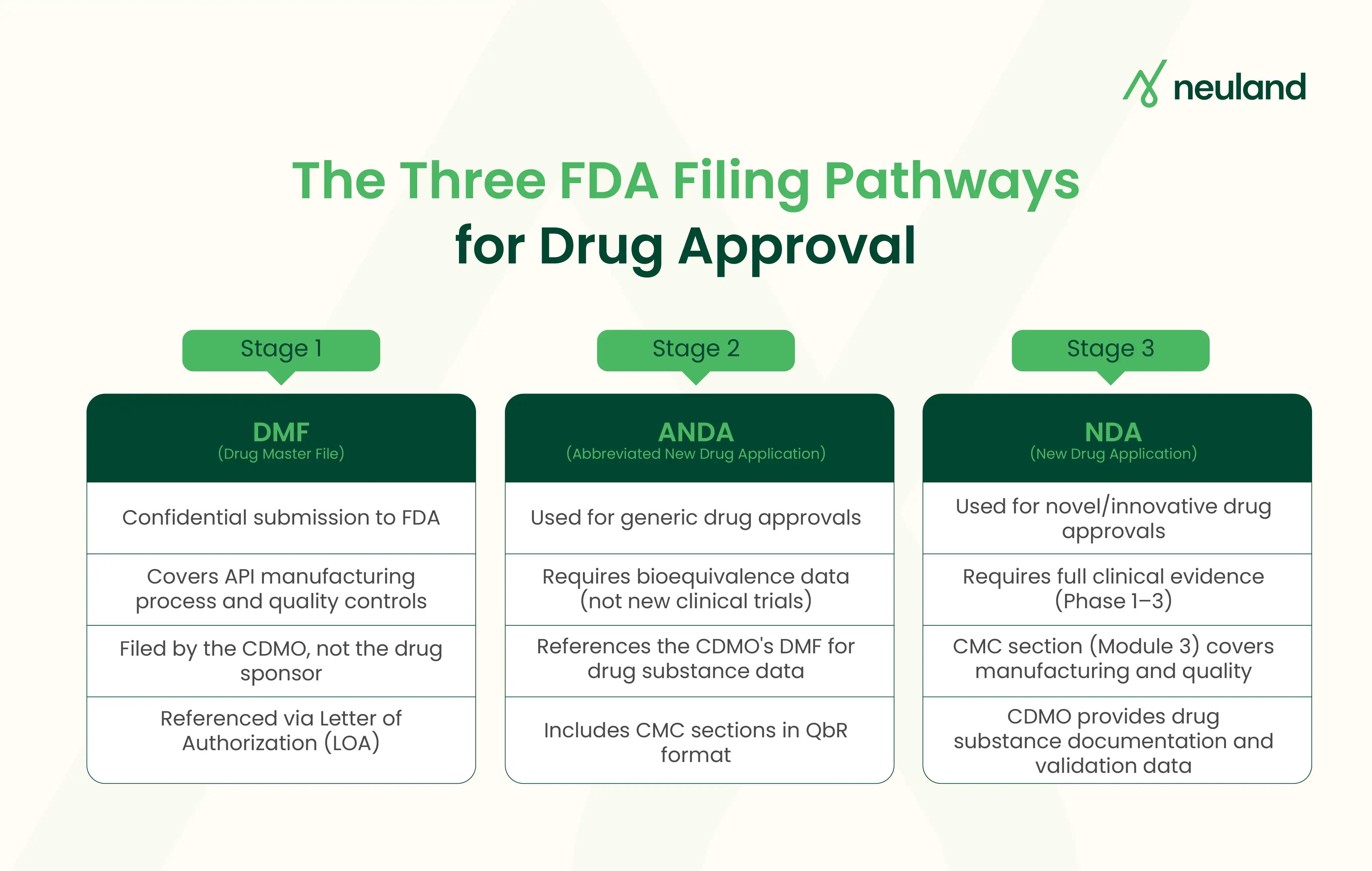
How Does a CDMO Support DMF Filing?
A Drug Master File is a confidential document submitted to the FDA. It holds detailed information about an API's manufacturing process, quality controls, and specifications. The FDA does not "approve" a DMF. It reviews the file only when a drug sponsor references it in their own application.
Here is what a pharmaceutical CDMO typically handles during DMF preparation:
- Synthetic route documentation covering each step of API production
- Analytical method validation for identity, purity, and potency testing
- Impurity profiling aligned with ICH guidelines (M7, Q3A, Q3D)
- Stability data under ICH long-term and accelerated conditions
- eCTD formatting for electronic submission through the FDA gateway
The CDMO also issues a Letter of Authorization (LOA) to each client. This LOA lets the FDA review the DMF in connection with the client's application. One DMF can support multiple sponsors. That makes it a scalable asset for any contract development and manufacturing organization.
The FDA now allows early DMF assessment six months before an ANDA submission. CDMOs that file proactively give their clients a clear time-to-market advantage.

How CDMOs Strengthen the ANDA Submission Process
An Abbreviated New Drug Application is the regulatory pathway for generic drugs. Unlike an NDA, it skips new clinical trials. Instead, the applicant must prove bioequivalence to the reference listed drug.
The CDMO's DMF forms the backbone of the ANDA's drug substance section. Beyond that, here is how CDMO services support the ANDA filing process:
- Manufacture exhibit batches at commercial scale for stability and bioequivalence studies
- Execute process validation to prove consistent production
- Prepare CMC sections using the FDA's Question-based Review (QbR) format
- Ensure inspection readiness for FDA pre-approval audits
Manufacturing deficiencies are a leading cause of Complete Response Letters. A CDMO with a clean inspection record — like one that clears a USFDA audit with zero 483 observations — directly reduces that risk.
Example: Consider a generic drug company filing an ANDA for a cardiovascular API. The CDMO's pre-existing Type II DMF already holds the process data and impurity profiles the FDA needs. The applicant simply references it through an LOA. This cuts months off the drug approval process.
The FDA approved 689 ANDAs in FY2025. Each one required solid drug substance documentation. In most cases, that documentation came from a CDMO partner.
| Ready to simplify your next regulatory filing? Work with a CDMO partner that brings proven CMC expertise and a strong track record in DMF, ANDA, and NDA support. Talk to a CDMO expert |
What CMC Support Do CDMOs Provide for NDA Submissions?
An NDA is the most detailed filing in the drug approval process. It covers a drug's full story across five CTD modules. Module 3, the CMC section, is where the CDMO's contribution lives.
For NDA submissions, a pharmaceutical CDMO typically provides:
- Registration batch manufacturing at commercial scale
- Drug substance documentation for CTD section 3.2.S
- ICH-compliant stability studies to establish shelf life
- Analytical method development and validation
- Technology transfer support from pilot to commercial production
- Pre-approval inspection preparation
The CMC drug development strategy usually gets locked in at the End of Phase 2 FDA meeting. CDMOs with NDA experience help sponsors prepare for these discussions. They know what data the agency expects and how to present it.
In 2025, the FDA approved 46 novel drugs. Many relied on CDMO partners for active pharmaceutical ingredient manufacturing and CMC filing support.
Why CMC Documentation Matters in Every Drug Development Filing
CMC documentation is the common thread across DMFs, ANDAs, and NDAs. It proves that a drug can be made consistently, safely, and at scale.
The challenges around CMC have grown significantly. Impurity management now involves multiple ICH guidelines. Scale-up from lab to commercial production often introduces unexpected variables. And regulatory expectations differ between the FDA, EMA, and PMDA.
This is exactly why drug companies turn to CDMO regulatory support for filing help. A CDMO that has filed dozens of DMFs and supported multiple NDA submissions brings deep institutional knowledge. Each deficiency letter answered and each inspection cleared builds expertise that individual sponsors rarely develop alone.
The global pharmaceutical CDMO market is projected to exceed $300 billion by 2030. Much of that growth comes from demand for regulatory CMC support.
Neuland Laboratories exemplifies the CDMO-regulatory partnership
As a focused API CDMO, Neuland Laboratories illustrates the scale and depth of regulatory support that specialized manufacturers provide. Neuland Labs supported by over 360 R&D scientists serving 500+ clients across 80+ countries
For pharma and biotech companies evaluating CDMO partnerships, the evidence points toward prioritizing regulatory expertise alongside manufacturing capability. Explore all the CDMO services.
FAQs
|
|
|
|
