From IND to Commercial: How Neuland Ensures Phase-Appropriate Development
The drug development process is inherently complex, with each stage bringing its own requirements and challenges. A one-size-fits-all strategy rarely works. Instead, companies must adopt a phase-appropriate development approach that aligns scale with the specific needs of each stage.
Early in development, speed and flexibility are critical. Methods and processes must be adaptable as knowledge evolves. As a molecule advances through the clinical trial phases, however, the focus shifts toward robustness, regulatory compliance, and scalability.
With more than four decades of experience as a global CDMO, Neuland Labs applies this phase-appropriate philosophy across small molecule and peptide programs.
From preclinical material preparation and IND-enabling studies to clinical phase manufacturing and commercial API production, Neuland supports development at every stage. The sections that follow explore how this approach is implemented from IND through commercialization.
Why Phase-Appropriate Development Matters
A phase-appropriate development strategy helps teams avoid two common pitfalls: over-investing too early or under-preparing for later stages.
Tailoring development activities to each phase helps organizations allocate resources better and de-risk the IND to launch transition. Regulatory expectations reinforce this phased model.
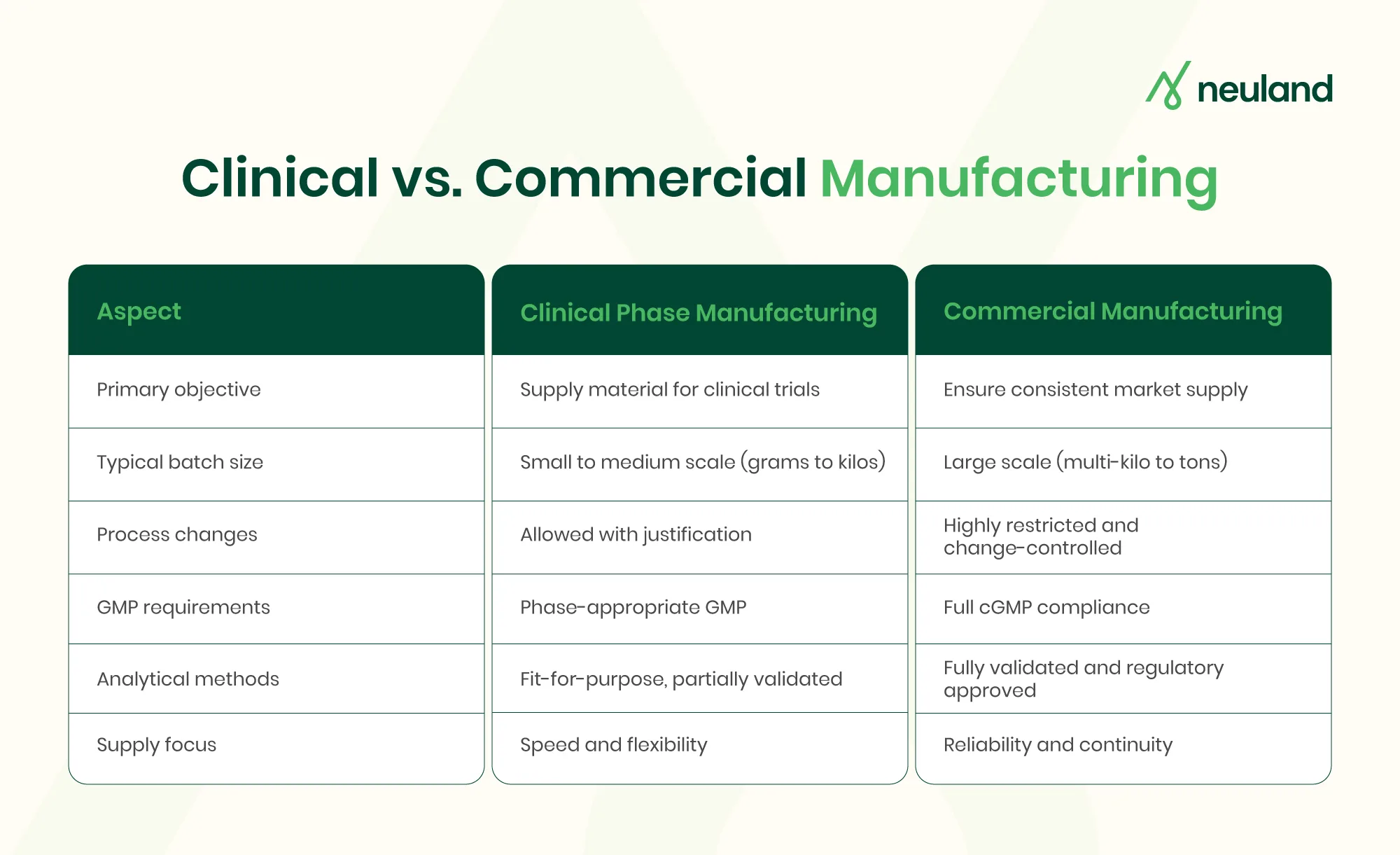
For example, regulators allow greater flexibility during early clinical development. Clinical phase manufacturing for Phase I trials is exempt from certain full Good Manufacturing Practice (GMP) requirements. Phase II and III programs are expected to operate under increasingly stringent GMP controls.
By the time a drug reaches NDA filing and commercialization, all manufacturing steps, analytical methods, and quality systems must be fully developed and validated. Adopting phase-appropriate drug development early makes the transition systematic, saving time, costs, and boosting regulatory readiness and supply reliability.
What Are Phase-Appropriate Solutions in Drug Development?
Phase-appropriate solutions in drug development align process maturity, analytical rigor, and regulatory effort with the specific needs of each development stage. The goal is to generate sufficient, decision-enabling data at the right time.
Early development: preclinical to IND / Phase I
In early development, speed and adaptability take precedence. The primary objective is to produce enough API of appropriate quality to support toxicology studies and first-in-human trials, while keeping processes flexible for change.
At this stage, Neuland focuses on essential CMC elements required for IND submission, without introducing unnecessary complexity.
Key phase-appropriate activities typically include:
- Fit-for-purpose process development to support small-scale API supply
- Material characterization, solubility, and preliminary stability assessment
- Early analytical methods qualified for reliability, not full validation
- Production of non-GMP tox batches or initial GMP clinical batches
Analytical methods are developed to ensure accuracy and basic specificity, acknowledging that both synthesis routes and methods may evolve. By limiting validation to what is required for Phase I, Neuland helps conserve API, time, and cost while maintaining regulatory acceptability.

Mid-stage development: Phase II and Phase III
As programs progress into Phase II and III, regulatory expectations increase, and supply demands grow. Phase-appropriate solutions now shift toward process optimization, scalability, and control. Neuland works with clients to refine chemistry, improve yield and purity, and demonstrate process robustness through pilot and engineering runs.
At this stage, development efforts typically expand to:
- Process scale-up and definition of critical process parameters
- Strengthened GMP controls and batch-to-batch consistency
- Phase-appropriate method validation aligned with ICH expectations
- Initiation of process validation and extended stability programs
Analytical methods move toward full validation using staged or matrix-based approaches, ensuring data readiness for regulatory submissions without unnecessary duplication. Manufacturing campaigns are executed under tightening GMP requirements, supported by Quality by Design (QbD) principles to manage risk and variability.
Neuland also accounts for operational realities such as fluctuating clinical demand and global trial requirements.
With flexible manufacturing capacity and facilities audited by major regulators, the company supports reliable clinical phase manufacturing across regions. By the end of Phase III, all CMC elements are fully developed, validated, and ready for regulatory review—precisely aligned with what authorities expect at this stage.
How Neuland Delivers Phase-Appropriate Solutions
Neuland Labs has built its services around a phase-appropriate approach, enabling seamless progression across the drug development lifecycle, particularly for complex small molecules and peptides.
As an integrated CDMO, Neuland provides continuity from early process design through scale-up, validation, and commercial manufacturing. The same teams that establish initial synthesis routes remain involved as processes mature, reducing handoff risks and preserving critical process knowledge.
This end-to-end engagement allows clients to anticipate phase-specific challenges early. Whether adjusting solvents for manufacturability or planning polymorph control strategies, Neuland’s phase-appropriate development effectively builds a scalable path to market from the outset.
Neuland’s peptide expertise further reinforces this model. Early discovery-scale synthesis can transition smoothly into clinical and commercial supply using a combination of solid-phase and solution-phase technologies.
Throughout development, Neuland Labs has deliberately evolved to match phase requirements, ensuring data quality without unnecessary delay or rework.
FAQs
|
|
|
|
