Analytical CDMO Services for API Development
Every API filing depends on one thing: data. Analytical data that proves your molecule is pure, stable, and well-characterized.
That's where analytical CDMO services come in. Testing, method development, regulatory documentation, and impurity characterization. All the analytical work pharma and biotech companies need before a molecule can file for approval.
How critical is this work? A 2025 Pharma Manufacturing analysis showed that 74% of FDA Complete Response Letters between 2020 and 2024 flagged quality or manufacturing problems. Weak analytical packages sit behind many of those rejections. These include unvalidated methods and gaps in impurity data. Also, stability profiles that don't hold up.
This article explains what these services actually cover, why they matter for regulatory outcomes, and what to look for in a partner.
What Analytical CDMO Services Cover in API Programs
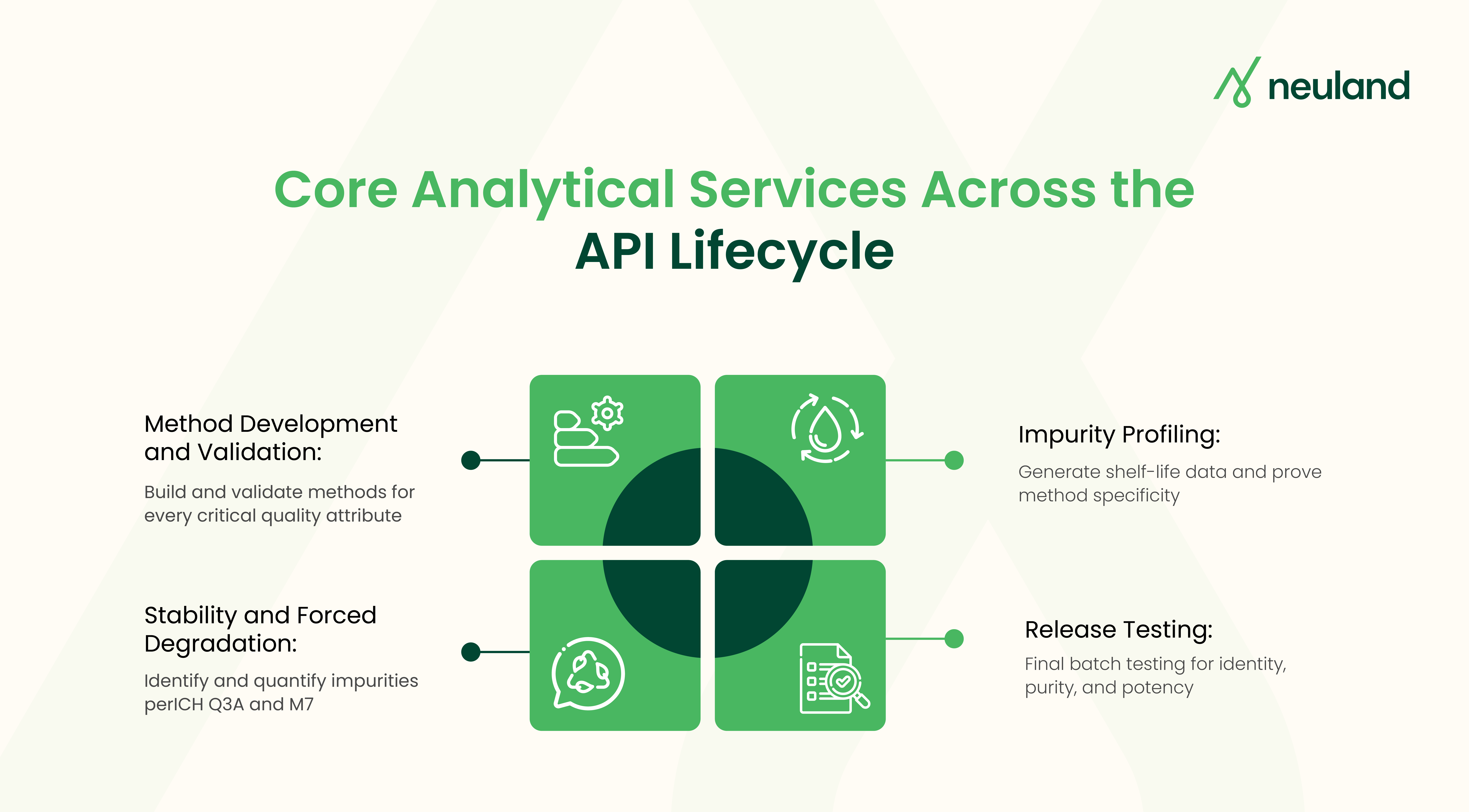
Analytical CDMO services touch every phase of an API program. The work starts when a process route is still being scouted and keeps going through commercial batch release. A typical scope looks like this:
Method Development and Validation
If you can't measure a quality attribute reliably, regulators won't accept it. Assay, impurity levels, residual solvents, water content, particle size. Each one needs its own validated method. ICH Q2(R2) sets the bar for what "validated" means: specificity, accuracy, precision, and documented proof that the method holds up under real-world conditions.
Impurity Profiling and Characterization
Regulators expect structural identification of every impurity above reporting thresholds. ICH Q3A sets these limits based on daily dose. Mutagenic impurities under ICH M7 demand even tighter controls, often at parts-per-billion levels.
Stability Testing and Forced Degradation
Stability studies under ICH Q1A generate the shelf-life data regulators need. Forced degradation studies confirm that analytical methods can detect relevant breakdown products. Both are required before filing.
Release Testing and Reference Standards
Final API batches need identity, purity, and potency testing before release. Reference standards require orthogonal characterization. These deliverables feed directly into Module 3.2.S of the CTD filing.
Why Analytical Quality Decides Filing Outcomes
Regulatory agencies have sharpened their focus on CMC data quality. FDA CDER warning letters jumped 50% in FY2025. Pre-approval inspections now target analytical labs directly.
The most common analytical deficiencies in recent filings include:
- Unvalidated methods that don't hold up under regulatory scrutiy
- Missing stability-indicating capability in HPLC methods
- Incomplete impurity qualification for unknowns above ICH thresholds
- Weak analytical bridging between clinical and commercial batches
These aren't theoretical risks. A single analytical gap can trigger a Complete Response Letter. That costs 9 to 12 months of lost market access.
Strong analytical CDMO services prevent these problems by building the analytical package in parallel with process development. Not as an afterthought.
How Analytical Work Connects to Process Development
The best analytical partners don't operate in isolation from manufacturing. They feed directly into process design through a Quality by Design (QbD) framework.
Here's how that works in practice:
- Critical Quality Attributes (CQAs) like purity, polymorphic form, and particle size are defined early
- Each CQA gets a validated analytical method that can detect meaningful changes
- Design of Experiments (DoE) studies use analytical responses to map process parameters
- The control strategy ties analytical data to manufacturing decisions at every step
This integration matters because methods developed on lab-scale batches often fail at commercial scale. New impurities appear. Retention times shift. A CDMO that builds analytical methods alongside process development catches these issues early.
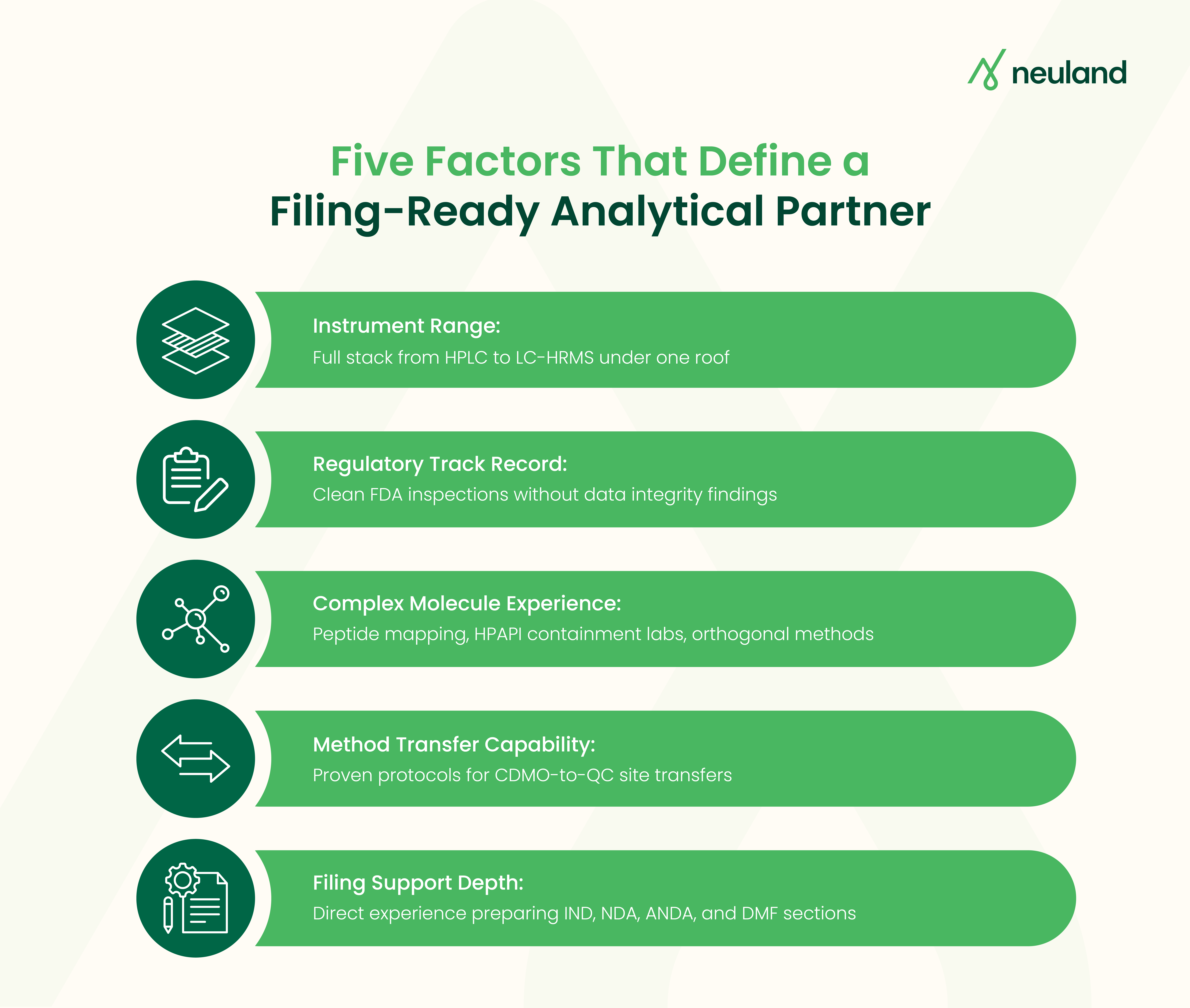
What to Look for in an Analytical CDMO Partner
Not every CDMO brings the same depth in analytical work. Here are the factors that separate strong partners from average ones:
- Instrument range. HPLC, UHPLC, GC, ICP-MS, Karl Fischer, XRPD, NMR, and LC-HRMS under one roof.
- Regulatory track record. A clean FDA inspection history without data integrity observations.
- Complex molecule experience. Peptide and HPAPI programs need orthogonal methods and containment-ready labs.
- Method transfer capability. Transfers to your QC site should happen without performance loss.
- Filing support. Direct experience preparing CMC sections for INDs, NDAs, ANDAs, and DMFs.
According to Grand View Research, the pharma analytical testing outsourcing market reached $8.96 billion in 2024. It's projected to grow to $14.56 billion by 2030. That growth reflects how many companies now treat analytical outsourcing as a strategic capability, not a commodity.
Building a Filing-Ready Analytical Package From Day One
The companies that avoid regulatory delays share one trait. They treat analytical development as a strategic decision, not a procurement task.
Neuland Laboratories brings this depth to analytical CDMO services for API programs. With in-house analytical infrastructure covering method development, impurity profiling, stability testing, and release testing, Neuland supports clients from early development through commercial-scale CDMO services. Their team works across small molecules, peptides, and complex APIs. Approvals from the FDA, EMA, and PMDA mean the data they generate is built for global regulatory review.
For teams planning an API program, the right analytical partner shapes the entire filing timeline. Talk to Neuland's team today.
FAQs
|
1. What do analytical CDMO services include for API development? Analytical CDMO services for API development include method development, method validation, impurity profiling, stability testing, forced degradation studies, release testing, and reference standard characterization. These deliverables feed directly into the CMC section of regulatory filings. |
|
2. Why do pharma companies outsource analytical work to CDMOs? Pharma companies outsource analytical work to CDMOs because maintaining a full lab with instruments like LC-HRMS and ICP-MS is expensive. CDMOs offer validated methods, filing experience, and instrument depth that most companies can't justify building internally. |
|
3. How do analytical gaps cause regulatory filing delays? Analytical gaps cause regulatory filing delays in a few common ways:
Between 2020 and 2024, 74% of FDA Complete Response Letters cited quality or manufacturing problems. Many traced back to exactly these kinds of gaps. Partnering with a CDMO that provides end-to-end analytical and manufacturing support from early development reduces the risk of finding these issues at filing. |
|
4. What should companies look for when choosing an analytical CDMO? When choosing an analytical CDMO, companies should look for broad instrument capabilities, a clean regulatory inspection record, experience with complex molecules like peptides and HPAPIs, proven method transfer protocols, and direct experience preparing CMC filing sections. |
