
Technology transfer from pharmaceutical companies to contract development and manufacturing organizations (CDMOs), is critical to successful drug development. When you consider the skills, knowledge, intellectual property, technologies, and manufacturing methodologies being transferred, it’s easy to comprehend both the importance and complexity of such an undertaking.
The expertise needed for an active pharmaceutical ingredient (API) technology transfer requires solid technical proficiency, a specific type of project management skill, commitment to innovation, regulatory knowledge, logical thinking, quality, compliance, adaptability, collaboration, continuous improvement and efficiency that assures R&D professionals, drug development scientists, and key decision-makers in the industry of success. This work is instrumental in shaping the future of drug development and manufacturing through strategic collaborations and advanced technological integrations.
API technology transfer requires a roadmap that clearly directs all the players, in particular the technology transfer team, from one stage to the next. Neuland’s tech transfer process includes the following:
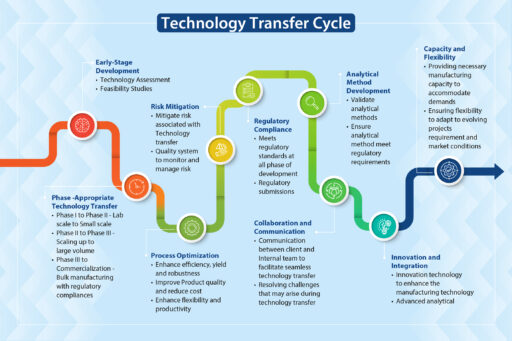
Stage One – Document the Scope and Objectives
The first step is to establish clear communication channels with the client. The synergy of collaboration developed early-on between different departments and teams is the path for a seamless transfer. It fosters a culture of trust and open communication that ensures every stakeholder, from the scientists to supply chain managers, is aligned with, and supports, the project’s goals and timelines.
Second, we agree on the scope and objectives of the project. Once the communication channels are established and trust built, it’s time to finalize the Scope and Objectives of the Technology Transfer. The client and CDMO achieve clarity on what they want and what is possible. There will be a give and take, especially once the baseline assessment is completed. Fortunately, a component of the communication channels includes regular updates and transparent discussions. Because of this process, all parties can adapt quickly to changing needs and expectations. Feedback and open dialogue facilitate the exchange of critical information, assuring every project is tailored to specific client requirements.
We then assess the client’s technology and processes. A detailed assessment identifies:
A proactive approach for the assessment means risk mitigation begins quickly – potential challenges are identified early on and we address issues before they become a problem and escalate.
Stage Two – Develop a Detailed API Technology Transfer Plan
The CDMO’s cross-functional project management team is responsible for creating a detailed tech transfer plan customized for each project. Our tech transfer plan includes:
Stage Three – Transfer Analytical Methods and Validate Methods
Stage One and Two focus on building the relationship with the client and creating plans to ensure a successful transfer. In Stage Three, we perform analytical testing by developing a comprehensive method transfer protocol outlining the objective, scope, and methodology for transferring and defining acceptance criteria including precision, accuracy, sensitivity, and specificity to prove that the knowledge is successfully transferred.
Method validation includes method demonstration by following regulatory guidelines (ICH Q2(R1)) and documentation which includes method protocols, validation protocols, raw data, results, and conclusions. Analytical method validation during the technology transfer process supports consistent quality and safety of the API at different sites.
Stage Four – Scale Up and Optimization
An effective approach to scale-up is anchored in optimized process development. Scientific expertise is foundational to the development of processes that are not only scalable but also efficient and cost-effective.
Advanced technologies are necessary to meet the growing and evolving needs in global healthcare. They need to be integrated into scale-up strategies. Adopting cutting-edge solutions enhances present manufacturing processes.
Your CDMO should focus on First Time-Right manufacturing. Unfortunately, this is not always emphasized enough. Rework is costly from both a financial standpoint and getting the product to market in a timely fashion. At Neuland, our goal is to avoid rework and streamline the path from development to production by making it right the first time.
Stage Five – Validation Studies
Needless to say, validation of processes and results is a critical component of API technology transfer. We follow robust protocols to determine if each step of the manufacturing process meets the necessary quality standards and is reproducible on a commercial scale. We conduct validation tests, thoroughly analyze the results, and complete a report including any and all corrective actions that must be taken.
Stage Six – Training and Knowledge Transfer
Training and the transfer of knowledge is a thread through all stages. When collaborating with a client on a complex technology transfer the transfer of knowledge starts on Day 1. In Stage Six, the training and knowledge transfer becomes more structured. Standard Operating Procedures are documented and taught. Quality procedures, concepts, and skills such as Continuous Improvement are documented and presented.
Stage Seven – Trial Batch and Verification
After the technology and knowledge is transferred it’s time to test whether the transfer is doing what it is supposed to do by running a trial batch of the product. The batch must be analyzed and verified that it meets the quality specifications and regulatory requirements. The ultimate question needs to be answered – is the API safe to scale up and go to market?
Stage Eight – Regulatory Submission
Meeting global regulatory standards is necessary if your product is ever going to get to market. An unwavering commitment to quality and compliance is at the core of a seamless, successful technology transfer process. Meeting and exceeding regulatory requirements while ensuring processes align with global standards is crucial. At Neuland, a focus on regulatory compliance is integrated into every phase of a technology transfer, from initial planning to final production, ensuring the highest level of integrity and excellence.
When the roadmap is assiduously followed by the cross-functional project management transfer team, they consider, implement, and meticulously document procedures throughout the stages. All procedures are designed to meet and exceed global regulatory standards. At this stage, the documentation is aggregated, formatted, and submitted.
Stage Nine – Continuous Improvement
Continuous improvement (CI) is the crux of maintaining a market lead. Continuously using feedback and data-driven insights to refine processes will maintain excellence. CI starts early, continues through the transfer process, and becomes on-going after the transfer process is complete.
Stage Ten – Technology Transfer Closure
Upon successful completion of all the stages and attaining compliance, the technology transfer cycle is closed.
Leveraging Digitalization for Seamless Tech Transfer, Data Integration
Digital transformation is revolutionizing our industry and new technologies are being used to enhance API manufacturing and technology transfer processes. The adoption of cutting-edge digital tools and technologies streamlines the transfer of information, optimizes processes, and improves overall efficiency from API development to production or additional manufacturing sites. Digital platforms allow high precision and control throughout manufacturing processes, ensuring compliance and consistent quality.
In addition to smooth communications among teams, digital technologies facilitate seamless date integration from various sources across R&D, process development and manufacturing ensuring transparency and real-time updates. These platforms have become a veritable gold mine for the industry improving data visualization and manipulation, increasing the amount of data readily used for decision making and standardizing formats. Collaboration between teams becomes easier and increasingly streamlined.
The ability of today’s process modeling and simulation powers reduces the need for extensive experimental trials to get the same information: an improved understanding of manufacturing processes, the ability to move and optimize parameters, and predict outcomes.
This digital approach expedites the tech transfer process and enhances the quality and reliability of outcomes at your CDMO.
Another proactive improvement is the use of Product Analytical Tools (PAT) to provide for real time monitoring visibility in manufacturing processes. These automated tools reduce manual labor and increase accuracy and reproducibility. Trouble shooting in real time, with ease, stops problems before they create waste.
Support for Your API Technology Transfer
At Neuland Labs, we have a long history of successful API technology transfers and possess a deep understanding of all of the elements of a successful technology transfer and its challenges. We have a proven track record of innovative solutions. Ask us about them!
We leverage our experience in technology transfer, digital transformation, risk management, compliance and robust quality systems, to assist your scale up endeavors. Our experience in this field gives us the ability to anticipate and prevent or solve problems of any complexity. Our goal: a scale up that meets your needs and runs smoothly each time.
Contact us to learn more about how we can help you.
 A company’s ability to pivot and adapt — its agility — is more than just an asset. In today’s dynamic contract manufacturing landscape, it’s a necessity. Agility allows a CDMO to respond and adapt quickly and efficiently to changes in the marketplace.
A company’s ability to pivot and adapt — its agility — is more than just an asset. In today’s dynamic contract manufacturing landscape, it’s a necessity. Agility allows a CDMO to respond and adapt quickly and efficiently to changes in the marketplace.
In fact, according to observations at the 2023 CPHI worldwide pharma industry event, flexibility in response to project demands— such as scope expansions, volume increases, project termination or adjustments in response to research outcomes—is considered the single most important characteristic of CDMOs.
Pharmaceutical contract manufacturers must always stay aware of and keep pace with ongoing supply chain disruptions, unprecedented technological advances, geopolitical instability, regulatory uncertainties, environmental changes and more. The nature of this current industrial reality makes agile manufacturing more important than ever.
This post explores five crucial ways in which agility helps your API CDMO partner respond to your changing needs – ensuring they have the flexibility and production capacity to scale and speed delivery of your products.
The Benefits of Agility in a CDMO
Agility supports competitive advantage, reduces downtime, and leads to significant cost savings, all of which drive profits for pharma companies and their manufacturing partners. Time saved means more support for further research and testing. And all of this ultimately paves the way to increased market share and higher profits.
Here are five ways agility drives speed to market.
Risk mitigation is based on a proactive approach to management that helps reduce downtime in the manufacturing process. Agile CDMOs can more easily mitigate risks related to market volatility. Since agile companies are by nature more productive and efficient, they can quickly adapt to market changes, supply chain challenges or changing customer needs and are well equipped to find new solutions to potential problems before they escalate into major issues.
Agile manufacturing systems are responsive by design. This means agile API manufacturers offer their partners optimized cost management and efficient resource allocation. This avoids the financial implications of process inefficiency. And more efficient processes not only increase productivity, they reduce waste, which also adds to cost savings.
Ideal contract manufacturing partners offer accelerated time-to-market. Rapid scaling of processes ensures timely product launches. This is critical in a highly competitive industry. Whether changes are being driven by consumer demand, technology, or market dynamics, an agile manufacturer can keep pace, delivering on time as needed.
Agile projects have been shown to reduce time to market by 55 percent or more. This reduction in time to market can lead to earlier realization of revenue, from which higher profits result.
Agile API contract manufacturers collaborate seamlessly with drug sponsor R&D teams in support of new drug formulations. This is essential as new drug formulations often require novel manufacturing processes and techniques.
Contract requirements can also change suddenly due to results of clinical trials and other research outcomes. Swift adaptation to research, development, and market changes is made possible by integrating emerging technologies and responsively iterating novel innovations and adaptations. The responsiveness of teams ensures effective collaborations that support timely product launches and efficient processes.
The regulatory environment of the pharmaceutical industry is constantly changing. The inherent responsiveness of agile processes makes them ideal for adapting to evolving global regulatory standards and requirements.
The ultimate end result of properly implemented agility is stakeholder satisfaction. Agile manufacturers are adept at ensuring a consistent and timely API supply, which boosts stakeholder confidence.
 Agility at Neuland Labs
Agility at Neuland Labs
At Neuland Labs, our strength lies in our ability to take swift actions and realign ourselves with market trends. Our commitment to decisive action has helped us steer our partners through 39 years of unpredictable market dynamics and helped us emerge as a leader in agile manufacturing.
Here’s how we help your drug product succeed:
An agile, adaptable mindset steers every decision we make. We rapidly respond to market dynamics, focus on flexible operations and partner needs to drive reliable speed, efficiency, and profitability for our customers.
Choose an Agile API Manufacturing Partner
When your API partner has optimized their manufacturing capacity for agility, you’ll benefit from increased organizational adaptability, collaborative capacity, rapid scaling, and accelerated time to market. Your choice of API partner must support your efforts to meet the needs of the customers you serve.
Have questions about how our agile manufacturing focus at Neuland Labs can help you meet your goals? Contact us to learn more.
Welcome to Part 2 of our series on managing API particle size. (You can find Part 1 here.) Today we’re talking about some of the new and emerging technologies that can impact API particle sizing. We also discuss in this post how Neuland successfully manages API particle size distribution projects.
As technology continues to develop, there are several new and emerging inventions and methods that can successfully and consistently impact and manage particle size for active pharmaceutical ingredients. Here are four methods which are not only showing promise but delivering on that promise.
Process analytical technology (PAT) such as focused beam reflectance measurement (FBRM) is one of the emerging technologies used to monitor the particle size and chord length distribution online during crystallization. When technology can successfully measure and manage crystallization, companies can speed up the work of creating APIs and get consistent results. Specifically, the Blaze Matrix PAT tool is useful for identifying polymorph transitions and morphology changes during crystallization.
Nanotechnology refers to manipulating atoms and molecules at nanoscale. Currently, many pharmaceutical contract manufacturers tackle the problem of low solubility by reducing the particle size of an API using micronization techniques. However, the demand for further improvements in drug dissolution has led to a shift from micronization to nanonization, as this technology significantly improves bioavailability and therapeutic efficacy.
Some techniques used to produce nanoparticles with precise particle sizes include high-pressure homogenization, nanoprecipitation, and spray-drying. At Neuland, we currently use homogenization and spray-drying to achieve lower particle sizes.
The in-line manufacturing process gives CDMOs the ability to conduct continuous and real-time quality assurance and control (QA/QC), which facilitates immediate adjustments during the manufacturing process. This ensures consistent particle size and improves the overall quality of the final product.
Artificial intelligence (AI) and machine learning (ML) are the newest techniques to manage API particle size. For example, a study published in Pharmaceutics included a list of ways AI and ML are currently being used in pharmaceutical product development.
One of the ways listed is using AI models for particle swarm optimization to optimize particle size distribution, dissolution profiles, and other formulation parameters. Another technique the study explores is artificial neural networks, which use AI/ML to predict the release behavior of active pharmaceutical ingredients (APIs) of various particle sizes under various conditions..
Neuland’s capabilities have helped our teams successfully manage several complex particle size distribution projects. Depending on the project and the materials, we use different strategies to make sure the particle size results in an end product with the characteristics desired by our customers.
In response to the growing need for tighter control over particle size, we launched a dedicated particle engineering lab. This lab contains facilities for crystallization development to achieve desired particle size and polymorphs for NCEs and APIs. It also contains particle size reduction equipment for both dry and wet milling as well as for micronization. The Neuland team has completed several projects that required a specific particle size and distribution. These successful projects necessitated the use of all three of the above-mentioned size reduction methods. We’ve also adopted the latest particle size engineering technologies, including implementing inline and online particle size meters as well as the latest crystallization techniques.
Neuland’s scientific team has conducted particle engineering studies using micronization for products such as Indacaterol maleate (D(90) less than 5 microns) and Ticagrelor (D(90) less than 10 microns). In another project, data generated from experiments with the compound Levetiracetam were used to optimize process conditions to meet the PSD requirement prior to kg scale production.
At Neuland, one of our unique strengths is our expertise in particle size reduction. Some of the particle size reduction techniques we use most often are the hammer mill, comill, multimill, wet mill, and air jet mill. Some products will have specific characteristics such as morphology, bulk density, and flowability crystal habit. Because of these characteristics during development, the lab will thoroughly study the suitable milling techniques and ensure that they can be optimized for scale-up.
We thoroughly study the solubility profile and generate the meta stable zone width (MSZW). Based on the solubility data we often perform various crystallization techniques such as cooling, evaporative, antisolvent, and reactive crystallizations to achieve the desired particle size. If crystallization does not provide the desired properties, then we use various size reduction techniques such as pin milling, hammer mill, comill, multimill, wet mill, and micronization to achieve the desired particle size, flowability and bulk density.
Have specific questions about how Neuland Labs can help you ensure the correct API particle size? Contact us to learn more.
API particle size is a key consideration in manufacturing quality pharmaceutical products, however, particle size is also notoriously difficult to control during manufacturing. Despite the difficulty, it’s essential to get right: particle size and morphology are the two most significant properties in oral solid dosage manufacture and performance, according to research published in AAPS PharmSciTech.
This post explores why API particle size matters and how API characteristics influence particle size. The next post in our series will highlight the emerging technologies for managing particle size.
In the pharmaceutical industry, particle size in APIs is a critical factor. This essential parameter has a direct influence on characteristics such as:
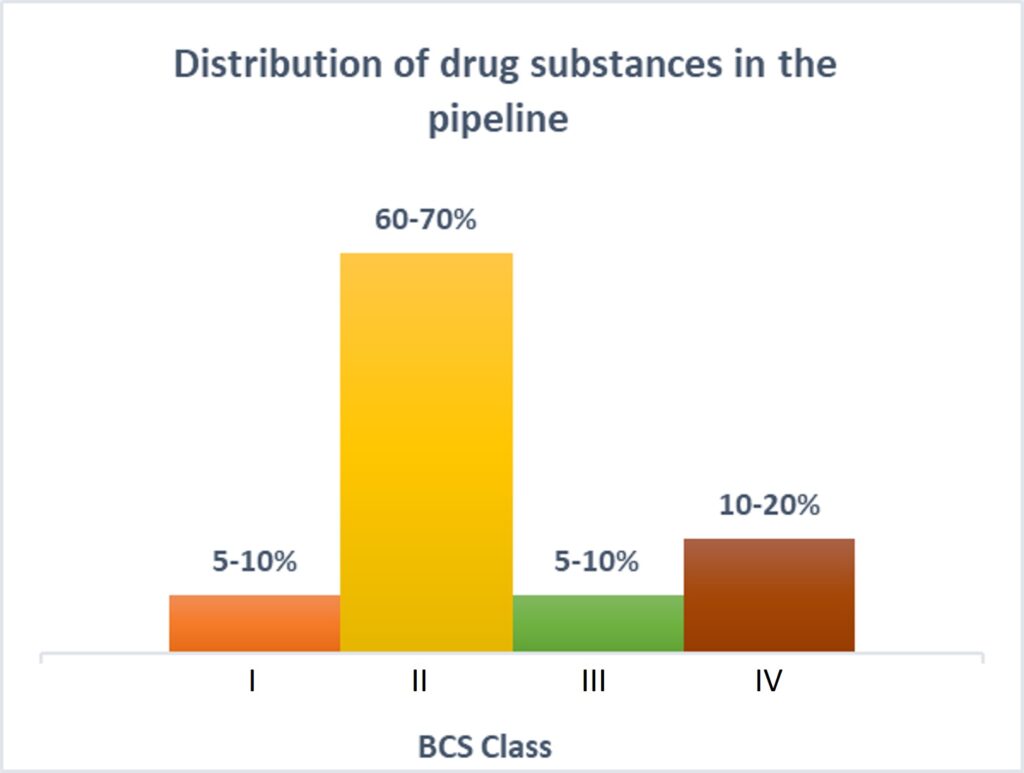 Those characteristics all affect the end patient, but API particle size also matters on the manufacturing side. API particle size, as well as particle size distribution, can affect the ease of the manufacturing process and the yield per batch.
Those characteristics all affect the end patient, but API particle size also matters on the manufacturing side. API particle size, as well as particle size distribution, can affect the ease of the manufacturing process and the yield per batch.
Another primary reason to have precise control over particle size is solubility. More than 80% of the new chemical entities (NCEs) in the current pipeline belong to BCS class II and class IV, making the future of pharmaceutical products look insoluble!
Particle size is an essential characteristic to creating a successful pharmaceutical product, but it’s a notoriously difficult aspect to control during the manufacturing processes. Even a small change in crystallization conditions, such as RPM/agitator/solvent ratios and cooling rates, can lead to significant changes in API particle size, as well as contribute to other property changes.
Another significant factor is changes in an API supplier, which can lead to new particle characteristics. If your company begins working with a new API supplier, be sure to assess the risks leading to changes in particle size. Carefully evaluate whether those changes will affect the drug properties, and if so, to what extent. If the new API size will affect drug properties, see if the method for characterization of particles is still valid.
Managing API particle size is one of the most difficult parts of the manufacturing process. API characteristics exert a profound influence on the strategies employed for managing particle size; therefore, different kinds of particle sizes may require different methods of handling. Here are some examples of characteristics of APIs that can help manufacturers manage the particle size carefully.
The solubility of an API dictates the bioavailability of a compound. APIs with low solubility demand a reduction in their particle size, thereby increasing their surface area and improving drug’s dissolution properties. To manage API particle size, manufacturers can use various reduction techniques, such as micronization, different types of milling, and nanosizing. These methods can be used to decrease particle size and increase solubility.
Crystalline structure plays a pivotal role in bioavailability. Amorphous APIs (devoid of crystal lattice) exhibit higher solubility and bioavailability compared to their counterparts. Therefore, preserving a desired amorphous state becomes critical and provides a precise control over particle size.
APIs with inherent chemical reactivity can pose unique challenges in maintaining particle size during manufacturing processes. Different conditions, such as lump formation during drying or a hygroscopic nature, can lead to the formation of an agglomerate, which frequently results in a higher particle size. To manage API particle sizes for chemically reactive compounds, manufacturers must control environments by using dehumidifiers and specialized equipment to ensure integrity of the final product.
 Managing the size of API particles is typically a complex process, but certain API characteristics are even more challenging to manage than others. Here are four characteristics that can make particle-sizing projects more difficult.
Managing the size of API particles is typically a complex process, but certain API characteristics are even more challenging to manage than others. Here are four characteristics that can make particle-sizing projects more difficult.
Many projects involve products with a single tier of PSD requirements. For example, a product may be d90 < 10 microns, which means that 90% of the particles are 10 microns or less. As with any pharma project, challenges can arise even during single–tier requirement products.
Projects become more challenging with two-tier requirements, in which materials are required to meet two distinct specifications. As the specifications move to three tiers, the difficulties become even more magnified. Controlling the particle sizes—for example, matching between the various tiers of d50/d90—can require the study of a combination of crystallization process and different particle reduction and sizing techniques.
Polymorphism refers to the ability of a compound to exist in multiple crystalline forms, known as polymorphs. Polymorphs have variations in physical properties such solubility rates, melting point, density, flowability, and particle size distribution. One polymorph may have larger particles or dissolve more readily compared to another.
Crystallization may also make an API more challenging. Crystallization challenges can involve precise temperature, solvent selection, and other parameters, thereby adding an extra layer of complexity to the process. Since crystallization can affect different pharmacokinetic properties, extensive studies are required to ensure the selected form does not change its particle size or stability over time. All these necessitate precise control over the manufacturing process to achieve consistent particle sizes.
APIs which are hygroscopic in nature can absorb water molecules. They are prone to moisture uptake from environmental air conditions leading to changes in the particle size. This can create agglomeration or clumping, causing non-uniform PSD. Moisture can also lead to the degradation or physical instability of the API.
Hygroscopic APIs are difficult to process for size reduction, as the materials tend to absorb the moisture and not have the flowability to pass through the mill. Manufacturers should ensure controlled environments with low humidity levels along with moisture-resistant packaging and desiccants to maintain desired particle size.
Stay tuned next month for part two of our series on API particle size. If you’re currently having challenges managing particle size and have questions, contact us here.
 Selecting the right pharmaceutical contract manufacturing partner for Phase 3 clinical and commercial supply is challenging. There are several critical qualifiers you need to consider:
Selecting the right pharmaceutical contract manufacturing partner for Phase 3 clinical and commercial supply is challenging. There are several critical qualifiers you need to consider:
Here’s how Neuland and Karuna Therapeutics partnered to meet the company’s clinical and commercial supply objectives.
Karuna Therapeutics is a biopharmaceutical company developing drugs for people living with psychiatric and neurological conditions. When Karuna and Neuland started discussions in early 2019, Karuna had an abbreviated timeline to scale up an NCE API, produce clinical trial quantities, and begin commercial manufacturing to support a combination CNS drug.
Tackling Challenges to Meet Phase 3 Clinical Trial Deadlines
Karuna and Neuland started working on the project in mid-2019. The scope of work included:
Tech Transfer: Translating Process Chemistry
It’s widely known in the industry that technology transfer can be a source of complex problems. CROs and CDMOs who focus earlier in the drug development cycle (preclinical through Phase II, for example) tend to be less familiar with process scaling issues that may arise at late-stage clinical and commercial scales, such as large scale hazardous chemistry. This is something that often is not taken into consideration during early R&D or process development. Raw material sources specified in early process design (discussed below) can be another potential pain point as procurement needs shift to bulk quantities.
Managing Supply Chain Risks: Backward Integration Via In-House Synthesis
Karuna – like other forward-thinking pharma companies – saw the need to have robust global Regulatory Starting Material (RSM) supply chains. Cost, delays, shutdowns, backed up ports, insufficient trucking capacity – the industry was surrounded by logistics risks and Karuna needed to ensure supply security.
Due to unforeseen challenges with the original RSM, Karuna and Neuland decided it would be advantageous for the client if Neuland developed capacity for RSM. By doing so, Neuland backward integrated production of the RSM, allowing for high quality and cost-effective manufacturing, and also established three domestic secure sources for the RSM’s starting material.
Handling Facility Shutdowns and Global Logistics
Shutdowns due to COVID outbreak were a particularly worrisome supply chain issue. When China closed, and India moved to follow suit, Karuna asked Neuland to ship available material to secure it in a GMP warehouse in the U.S. for its ongoing clinical trials.
At the time, global logistics were in a state of chaos, but Neuland’s expert logistics team ensured sufficient material was pre-positioned in the U.S. to meet the Company’s immediate clinical needs.
Complex, Hazardous Chemistry at Scale While Improving Yield and Speed
From a process design standpoint, handling large volumes of sodium cyanide poses risks. The route development team at Neuland was able to eliminate multiple hazardous reagents and large-scale reaction conditions while increasing yield and reducing cost.
The original process design produced low yields at each of its four stages. A cross-functional team at Neuland evaluated this 4-stage process, identified potential gaps, and created greener, more efficient and cost-effective processes.
Other Project Highlights Included:

Comprehensive Team Approach
Quickly approaching timelines required frequent collaboration from Neuland and Karuna, including process chemistry, analytical development, process engineers, Quality, EHS, production, and export logistics teams. A collaborative approach ensured seamless information-sharing which accelerated and simplified scaling.
Given the global and process-related complications – the project advanced swiftly.
Successful Design, Development and Launch
While API projects often feature one or two particularly strong outcomes, this project hit nearly every high point imaginable – from successful scale-up on abbreviated timelines, improving cost efficiencies, meeting regulatory objectives, and improving the client’s (and our own) supply chain security.
Contact us to learn how Neuland can make your next NCE API project a success, write to Saradhi@neulandlabs.com
 Specialty drug APIs are both a blessing and a challenge for pharmaceutical companies. They are essential for treating patients and saving lives. The demand for them is high, which promises revenue growth. However, even in the extremely lucrative market of APIs — the API market was valued at around $228.5 billion in 2022 and is expected to expand at a 6.5% CAGR through 2023 — the hurdles around specialty drug APIs remain. The high demand has drawn many companies to invest in these products, but many find they aren’t prepared to properly scale specialty APIs due to the difficulties in the process.
Specialty drug APIs are both a blessing and a challenge for pharmaceutical companies. They are essential for treating patients and saving lives. The demand for them is high, which promises revenue growth. However, even in the extremely lucrative market of APIs — the API market was valued at around $228.5 billion in 2022 and is expected to expand at a 6.5% CAGR through 2023 — the hurdles around specialty drug APIs remain. The high demand has drawn many companies to invest in these products, but many find they aren’t prepared to properly scale specialty APIs due to the difficulties in the process.
In this article, we explain some of the common challenges that drug companies face when working with specialty drug APIs, as well as some ways to mitigate those challenges with the right contract manufacturing partner.
Specialty drug APIs have high complexity and diversity of chemical structures and synthesis routes. Companies must have a team with deep expertise and experience in organic chemistry, analytical chemistry, process development, and scaling to manufacture these products. Even if a company can find the right talent, the synthesis is still complex. Each drug is different and requires a unique, detailed mapping of how process conditions may affect product quality.
One effective approach to tackle this complexity is to outsource development, scale-up, and manufacturing to a contract manufacturer who specializes in these kinds of drugs. Collaborating with an organization such as Neuland, which is equipped with a dedicated state-of-the-art R&D center with around 300 scientists skilled in complex chemistry and process development, ensures your organization has access to the requisite expertise and resources for success.
Another challenge facing pharmaceutical companies is the stringent quality and regulatory requirements for specialty drug APIs. These drugs demand rigorous testing, validation, documentation, and compliance with global standards such as USFDA, EMA, PMDA, etc.
The regulatory and quality requirements become even more complex for global companies. Different countries and regions have separate guidelines about the manufacturing of APIs and highly active drugs (for example, Europe’s EMA, China’s CFDA, Mexico’s COFEPRIS, USFDA, Canada’s Health Canada, Brazil’s ANVISA, and India’s CDSCO).
ActaBiomed, a clinical medicine journal, published an article that suggested that the best way to overcome these regulatory challenges is to standardize requirements across countries as well as across drug types. Many regulatory bodies use words like “some” and “certain” when describing which regulations apply to which kinds of chemicals that must be manufactured separately. Harmonization among countries and establishing a common scientific language could significantly increase manufacturing efficiency and ease regulatory confusion.
Until then, how can drug sponsors manage the complexities of global regulatory bodies? One solution is to work with a manufacturer, such as Neuland, with expertise in specialty drug APIs. Manufacturers with this expertise can synthesize, manufacture, and supply APIs and advanced intermediates for specialty drugs, especially those that target rare or orphan diseases. These companies can also offer their customers cost-effective synthesis, IP protection, and regulatory support.
 The high cost and risk of developing and manufacturing specialty drug APIs is another challenge for pharmaceutical companies. These APIs often require significant investments in R&D, infrastructure, and equipment. Even when an organization successfully develops a specialty drug API, there are still many expensive risks, such as:
The high cost and risk of developing and manufacturing specialty drug APIs is another challenge for pharmaceutical companies. These APIs often require significant investments in R&D, infrastructure, and equipment. Even when an organization successfully develops a specialty drug API, there are still many expensive risks, such as:
Two recommendations on how pharmaceutical companies can address risk and cost challenges include:
Neuland minimizes risks with our dedicated mini plant for scaling up new products. We also have a process safety lab for evaluating the hazards of chemical reactions. Neuland conducts safety studies to assess the potential risks associated with the manufacturing processes used to produce drug substances and to implement appropriate measures to mitigate these hazards and ensure the safety of workers, the environment, and the final product.
Development of specialty drug APIs is also hampered by the intense competition and innovation in the Specialty Drug API market. The industry requires constant monitoring of the latest trends, technologies, and opportunities for differentiation and value addition.
One contributing factor to the fierce competition is the increasing complexity of novel targeted therapeutics and the lack of in-house manufacturing expertise. According to researchers at Imperial College London, this has led to an increase in mergers and acquisition (M&A) and outsourcing strategies, making it harder for smaller companies and startups to succeed against global corporations with more funding.
Neuland’s core values help our clients succeed in this competitive environment. These values include customer-centricity, reliability, accountability, ownership, openness, and transparency. We also offer a no-compromise policy of not competing with our customers for finished products.
Contact us today to see how Neuland can help you overcome barriers and launch a successful specialty drug API product.
To be successful, pharmaceutical innovators and biotech organizations need to optimize production and achieve higher yields of their active pharmaceutical ingredients to make products more cost-effective. To realize these results, proper scale optimization is vital.
To learn more about how scale optimization projects unfold and how they can be achieved most effectively, we sat down with Dr. Mahender Rao Siripragada, president of R&D at Neuland Laboratories.
A: Basically, scale optimization involves improving and optimizing the manufacturing process of pharmaceutical products, with the goal of increasing production efficiency, reducing costs, and ensuring consistent product quality.
Key aspects of every project include a comprehensive analysis of the existing manufacturing process, an assessment of scale-up considerations, process optimization to maximize productivity and minimize variability, technology evaluation, risk assessment and mitigation, validation and regulatory compliance, implementation, monitoring, and strategies to ensure continuous improvement over time.
In addition, there’s a lot of multi-departmental collaboration throughout the entire project. Our cross-functional teams include process engineers, scientists, quality control experts, regulatory specialists, and manufacturing personnel to ensure success.
A: Process research and design are the most critical stages because they ensure the development and manufacturing of high quality, safe and effective drugs.
Process research involves exploring new technologies and methods that can be used to manipulate chemicals in compounds to create new drug products. This may focus on developing new methods of synthesis or improving existing manufacturing processes.
Process design, on the other hand, involves taking the results of the process research and designing processes that can efficiently and effectively produce the API on a large scale. That involves selecting appropriate equipment, defining critical process parameters, and setting quality standards that ensure that each batch produced is a high quality during the process research phase. Chemists and researchers work together to understand the chemistry of the compounds and identify potential routes to synthesis. They also use computer modelling laboratory experiments.
Another approach is to enhance the synthesis process to ensure that it is safe and effective. Process engineers and chemists work together to design the manufacturing processes for the API determine where it can be optimized and establish critical process parameters — the points in the manufacturing process that have the most significant impact on quality. These must be carefully controlled to ensure the safety and efficacy of the drugs produced.
A: That depends on the specific drug product being produced and the scale of production required. If the current process is inefficient or produces a low yield, it may require significant optimization. On the other hand, if the manufacturing process is already fairly efficient, the scale optimization may be more focused on fine-tuning the process to improve yield or reduce costs. Scale optimization can also be an ongoing process as new technologies and methods are developed.
A: Route Scouting Scale optimization is about making an existing active pharmaceutical ingredient manufacturing process more efficient. It aims to improve the process so that it yields the desired outcomes while minimizing costs and environmental impact, as well as ensuring that the produced batch meets regulatory requirements.
on the other hand, is focused on identifying new ways of synthesizing an API. This happens in the early stages of development when researchers aim to determine the most efficient and cost-effective process. In route scouting, various options are proposed and explored to identify the most optimal way.
In summary, scale optimization focuses on improving an existing process, while route scouting aims to find a new and better process. In both cases, the goal is to ensure the optimization of manufacturing processes, resulting in high quality drugs that meet regulatory requirements.
A: There are quite a few. I’ll try to summarize them quickly.
At the top of the list are process control and quality assurance. These are crucial to maintaining consistent quality in every pharmaceutical manufacturing process. Developing robust process control strategies and implementing quality assurance measures throughout the chemical process development are demanding but essential tasks. They include monitoring critical process parameters, implementing analytical techniques, and ensuring batch-to-batch consistency.
Ensuring the safety of the chemical processes and compliance with regulatory requirements are also critical challenges. Pharmaceutical companies must adhere to strict quality standards, Good Manufacturing Practices (GMP), and environmental regulations. It’s our job to ensure they can demonstrate the safety and efficacy of the processes and products to regulatory authorities.
Moving from laboratory-scale to large-scale production is another significant hurdle. Every process must be scalable to meet the demand for commercial production while maintaining product quality and consistency. Factors such as equipment design, raw material availability, and process control need to be considered during scale-up.
Then there’s time to market. The pharmaceutical industry is highly competitive, and getting new drugs to market quickly is essential. Developing and optimizing chemical processes within tight timelines requires efficient project management, collaboration between different teams, and effective communication.
Cost is always a major concern. Developing new chemical processes for pharmaceutical manufacturing can be expensive. In the first place, the industry must invest in research and development, equipment, facilities, and skilled personnel. The cost of scaling up a process from the laboratory to commercial production can also be significant.
Developing efficient and high-yield chemical processes is essential in order to optimize production and minimize costs. Pharmaceutical companies aim to maximize the yield of the desired product while minimizing waste and by-products. This requires expertise in process optimization, reaction engineering, and separation techniques.
New chemical processes involve intellectual property considerations. Pharmaceutical companies invest significant resources in research and development, so protecting their proprietary knowledge and innovations is crucial. They must navigate patent laws and ensure that their processes remain confidential and secure.
Last but certainly not least is environmental impact. Sustainability and reducing the environmental footprint of pharmaceutical manufacturing is gaining increasing importance. There’s growing demand to develop chemical processes that minimize waste generation, energy consumption, and the use of hazardous materials. Companies need to explore greener alternatives and adopt environmentally friendly practices.
Addressing these challenges requires a multidisciplinary approach, involving chemists, engineers, regulatory experts, and other professionals to overcome technical, regulatory, and operational hurdles. Collaboration between industry and academia can also play a vital role in tackling these challenges and advancing the field of pharmaceutical process development.
A: Neuland’s vast technical and scientific expertise in route scouting is helping our partners identify the best, most feasible and cost-effective routes of synthesis for the API they want to manufacture. We bring strong experience in supply chain management to the process, which helps identify the best options for potential vendors. Our dedicated analytical team enables feasibility studies to be completed very quickly.
The advantages of strong technical expertise, seamless coordination between cross-functional teams, along with the quality policies adopted by our management have established Neuland as one of the best API/CMS manufacturers in the world.
Contact us today to discuss your next scale optimization or route scouting project.
The development of drug substances heavily depends on a thorough and precise understanding of its different processes. One of these critical steps is crystallization, which has a significant impact on the properties of active pharmaceutical ingredients (APIs). By optimizing the approaches to crystallization, we can improve the stability, solubility, and bioavailability of APIs, which are essential factors in the successful development of effective drugs.
What is Crystallization?
Crystallization refers to the transformation of molecules or atoms from a liquid or gaseous state into a solid state. This process results in the formation of crystals, which are pure substances with atoms arranged in a well-defined and rigid crystal lattice to minimize their energy state.
The properties of the solid state, including the size, shape, and internal structure (morphology) of the crystals, can have a significant impact on the performance of a drug.
In drug development, various types of crystallization processes are utilized, such as cooling, evaporation, and reactive crystallization. Each of these methods offers distinct advantages. The selection of the appropriate process depends on factors such as the nature of the drug, required purity levels, and specific physical and chemical properties of the active pharmaceutical ingredient (API).
Challenges in Crystallization
Crystallization, an essential aspect of pharmaceutical manufacturing, presents a range of obstacles. Its complex nature, combined with the need for precise outcomes, demands specialized expertise and resources.
Consistency is vital for the development of active pharmaceutical ingredients (APIs). It is necessary to achieve consistent results in successive crystallization runs conducted under identical conditions. However, due to the process’s sensitivity to even slight variations in factors like temperature, supersaturation levels, and stirring rates, maintaining reproducibility is often difficult.
To achieve reliable results, your contract manufacturing partner must possess expertise in the crystallization process. This expertise enables them to select the appropriate process parameters, facilitating consistent production of crystals with the desired size, shape, and purity while minimizing downstream processing issues.
The shape and size of crystals, known as crystal morphology, can significantly impact a drug’s physical and chemical properties, including solubility, stability, and bioavailability. However, predicting and controlling crystal morphology is a complex task that necessitates a profound understanding of the solute-solvent interaction, nucleation and growth rates, and the conditions of the crystallization process.
Even trace amounts of impurities can disrupt the crystallization process, leading to issues such as alterations in crystal structure, hindered crystal growth, and even failed crystallization. Controlling impurities originating from both the solute and the solvent is a crucial challenge in pharmaceutical crystallization.
Laboratory-scale crystallization often does not seamlessly translate to larger-scale production due to variations in parameters like mixing and heat transfer. The scale-up process typically requires iterative adjustments and is time-consuming, as careful optimization is necessary to maintain product quality and process efficiency.
Polymorphism refers to a compound’s ability to exist in multiple crystal structures, each with distinct physical properties. While polymorphism offers opportunities for optimizing drug properties, it also presents challenges in controlling which polymorphic form is obtained during crystallization. Undesired polymorphic transformations can occur during storage or processing, negatively impacting the drug’s properties.
Crystallization Process Steps
Each step in the complex crystallization process plays an essential role in shaping the final product.
Selection of the appropriate solvent is the key first step in the process. You must consider solubility and safety when choosing the proper solvent.
Crystallization begins once supersaturation, a state where the concentration of the solute surpasses the solvent’s ability to dissolve it under specific conditions, is achieved. This imbalance is typically attained by manipulating temperature, reaction, evaporation, or pressure to exceed the solute’s natural solubility in the solvent. The delicate dance of this manipulation is integral, as the degree of supersaturation influences both the rate of nucleation and crystal growth that follows.
Following supersaturation, the solution undergoes nucleation. Here, solute molecules or atoms dispersed in the solvent start to congregate into stable clusters, forming a nucleus. This aggregation is the seed from which a crystal will grow. Precise control over nucleation is crucial, as the number of nuclei formed will dictate the number of crystals and inversely affect their size.
Once the nucleus forms, the stage of crystal growth commences. Additional solute molecules continue to deposit onto the existing nuclei. As they arrange in a defined, repeating pattern, the crystal begins to grow.
This stage is key to determining the crystal’s size, shape, and quality. Several factors can influence this growth, such as temperature, agitation, and the rate of supersaturation reduction. Striking a balance between development and nucleation is often the key to achieving the desired crystal size distribution.
The final step in the crystallization process is removing and isolating the crystals from the remaining solution. This isolation is typically accomplished using filtration or centrifugation, followed by a drying step to remove residual solvent. It’s important to handle the crystals gently during these steps to prevent damage or alteration to their carefully grown structures.
Common Crystallization Parameters and Transformations
Understanding the parameters involved in crystallization is vital for process control and optimization. Some of the common parameters include supersaturation levels, temperature, and pH. Monitoring and managing these parameters helps ensure optimal conditions for crystal formation.
Transformations are changes in solid forms during or after crystallization. They could involve polymorphic transformations (changes in crystal structure), amorphous to crystalline transformations, and desolvation (loss of solvent from solvate crystals).
Neuland’s Expertise in Crystallization
Our research team at Neuland focused on enhancing the physicochemical properties of Voxelotor, an API utilized for treating sickle cell disease. Voxelotor is a Class II drug with poor solubility. To address this issue, we employed crystallization techniques, resulting in significant improvements.
The outcomes of our study, titled “Physicochemical aspects and comparative analysis of Voxelotor and its salt and cocrystal,” were recently published in the Journal of Molecular Structure. The cocrystal of Voxelotor with Succinic acid exhibited notable enhancements in solubility and stability compared to the free base form. This work highlights our unwavering commitment to advancing drug development by employing innovative crystallization approaches.
Crystallization impacts multiple aspects of a drug’s quality and performance. Navigating the complexities of this process requires a deep understanding of crystallization chemistry, expertise in process control, and the ability to solve inherent challenges.
Contact us today if you want to improve stability, solubility, and bioavailability.
 There have been significant changes in the pharmaceutical industry over the past few years, and some of them have led to significant breakthroughs. One example is the growth of deuterated chemistry in the biopharma space.
There have been significant changes in the pharmaceutical industry over the past few years, and some of them have led to significant breakthroughs. One example is the growth of deuterated chemistry in the biopharma space.
With demand soaring for new drugs and medications, deuterated drugs can address a wide variety of problems at an affordable price. Deuteration chemistry has become a popular emerging frontier with tremendous potential to provide medications with improved pharmaco-kinetic and toxicological profiles.
What do pharmaceutical companies and product managers need to know about deuterated drugs, and how could they help you improve your processes?
What Is Deuteration Chemistry?
First, it’s important to discuss the meaning of ‘deuteration chemistry.’ It refers to using deuterium instead of hydrogen in the molecular structure. Hydrogen, the first element on the periodic table, has one electron, one proton, and no neutrons. Deuterium is similar to hydrogen in that it has one proton and one electron, but it also has a neutron. This makes deuterium an isotope of hydrogen, but the extra neutron adds some additional weight to the atom. Deuterated drugs exhibit superior pharmaco-kinetic or toxicological properties due to stronger deuterium–carbon bonds.
The incorporation of deuterium further leads to increased chemical stability, which is reflected by its slower rate of metabolism in the human body when ingested.
What Are the Advantages of Deuterated Drugs?
 There are some significant advantages of deuterated drugs. One of the immediate benefits is that deuterated drugs typically possess a much longer half-life (e.g., the amount of time it takes the body to metabolize the medication).
There are some significant advantages of deuterated drugs. One of the immediate benefits is that deuterated drugs typically possess a much longer half-life (e.g., the amount of time it takes the body to metabolize the medication).
The C-D bond is approximately 10 times stronger than the C-H bond, so it takes much longer for the body to break down the medication. A deuterated drug is much more resistant to enzymatic cleavage, which means that deuterated drugs will stay in the body for a longer amount of time.
Some of the main advantages of a longer drug half-life include:
Deuterated drugs can reduce expenses during the research process and help bring new medications to the market more quickly.
Traditional drugs with a C-H bond can fail testing for a variety of reasons, including:
Deuterated drugs can sidestep these issues because they are more efficient and less prone to side effects. They also have access to streamlined approval processes with the FDA. Because many of these issues can be avoided with stable deuterated drugs, clinical trials tend to focus almost exclusively on pharmacokinetics and toxicity, streamlining the approval process and helping companies get new medications to the market more quickly.
Where Does Deuterium Come From?
Clearly, there are a lot of advantages that deuterium can provide in the world of pharmaceuticals, but where exactly does it come from? It is true that hydrogen is significantly more common in the environment than deuterium, so deuterium enrichment is a vital part of the manufacturing process. Heavy water (D2O) produced through the deuterium enrichment is the most convenient source of deuterium.
We manufacture advance deuterated reagents, starting from heavy water (D2O) and deuterated methanol. This process has multiple stages, and we combine the result with the appropriate API to provide our clients with the target compound.
Neuland and Deuteration
Because of the stronger carbon deuterium bonds, deuterated drugs often demonstrate superior pharmacokinetic and toxicological properties. A handful of deuterated drugs have been approved, and many clinical trials continue to move forward.
Neuland Labs is proud to be an industry leader in this developing niche, and is a commercial supplier of deuterated APIs. If you would like to learn more about how we can help you take advantage of deuteration chemistry to improve a therapeutic API, contact us today.
 Respected Irish business Niall FitzGerald once said, “Sustainability is here to stay or we may not be.” A supporter of sustainable business initiatives in Europe, he has also pointed out that capital markets are mass migrating towards higher ESG scores.
Respected Irish business Niall FitzGerald once said, “Sustainability is here to stay or we may not be.” A supporter of sustainable business initiatives in Europe, he has also pointed out that capital markets are mass migrating towards higher ESG scores.
The pharma industry is one of the world’s largest, accounting for nearly $1.5 trillion in revenue in 2021. It has an equally large environmental footprint, consuming enormous volumes of water, and generates chemical and material waste. Pharma is also 55% more emissions-intensive than the auto industry. As much as 80% of this consumption has been attributed to manufacturing operations.
To truly achieve sustainability, the industry will need to rethink and transform manufacturing processes – from product design to distribution of goods.
If you’re thinking “yes, we need to get started doing this,” here’s some good news: the push to discover new approaches is already well underway – both here at Neuland and in the broader drug development and manufacturing industry. Breakthroughs – like the examples below – are often refined, adapted, examined and re-examined… some may become established SOP. These three examples impact areas critical to the pharma industry, spanning energy consumption, resource utilization and waste reduction.
According to Fierce Pharma, global consulting firm Oliver Wyman reports that pharma companies are making commitments to environmental sustainability, but practical barriers make it difficult to deliver on that commitment.
Pharma is Growing. Resource Requirements Need to Shrink.
As the drug industry continues to grow in size and volume, the industry’s emissions and resource usage are growing alongside it. Managing increasing energy consumption and environmental pollution at scale are no longer simply business continuity issues, they have become existential challenges. At scale, improving process design efficiency has across-the-board implications – potentially lowering waste and effluent production, energy requirements, water usage, resource extraction and more. Selectively employing single-use systems, for example, can make sense – depending on batch sizes and end-target product volumes.
Neuland Labs is proud to be a part of the journey discovering the right balance of measures the drug manufacturing industry can take.
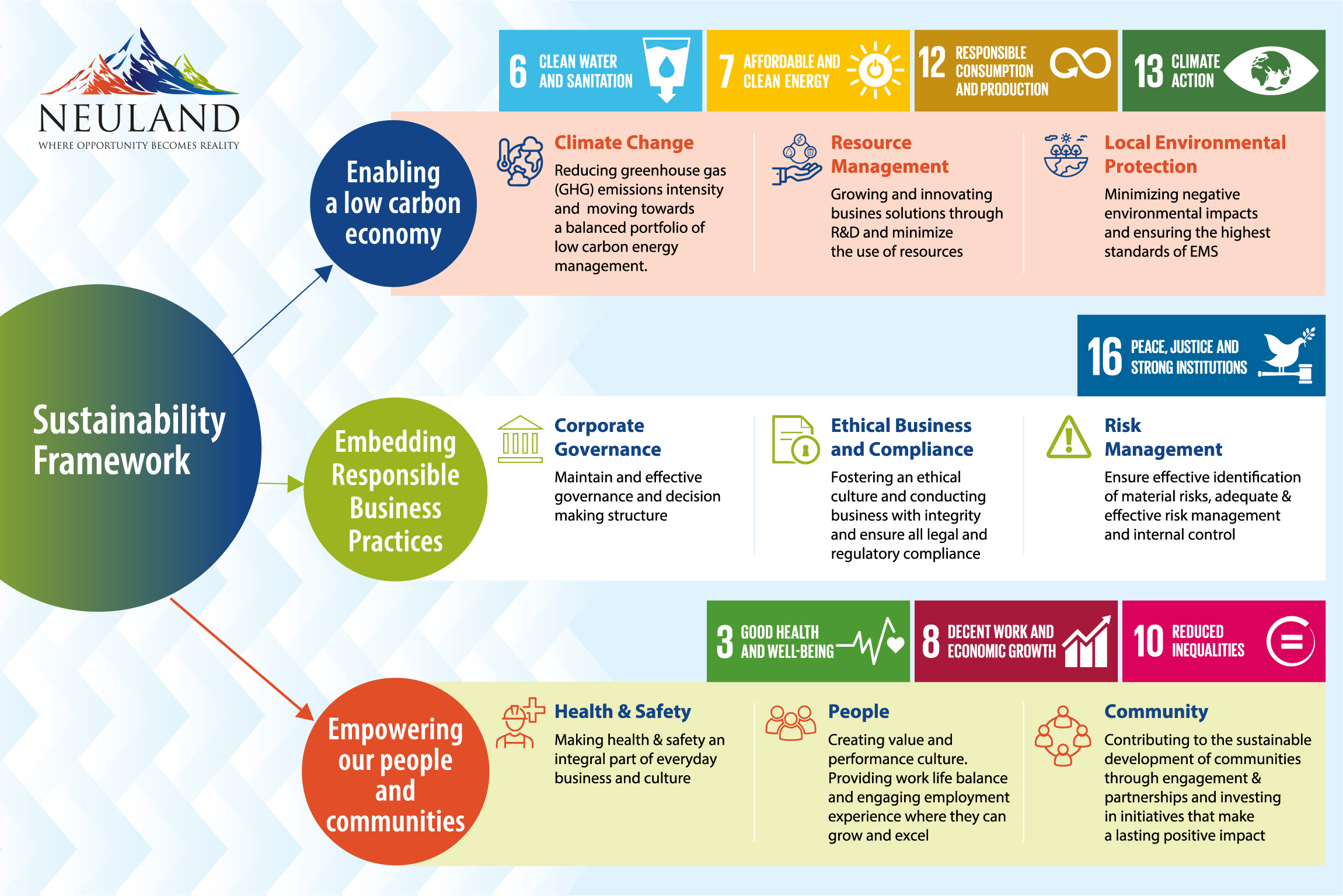
Emphasizing Sustainability at Neuland Labs
Sustainability and environmental health and safety have been a focus at Neuland for years, but recently our commitment to sustainability was formalized among senior leadership as a guiding principle for our strategic priorities.
Communication with stakeholders has been – and will continue to be – critical to our efforts, spanning investors, board members, workers, clients, suppliers, the community and regulators.
According to Dr. Davuluri Rama Mohan Rao, Executive Chairman of Neuland Labs:
“By obtaining insights from 125 of our stakeholders across different sectors, we identified six key themes of sustainability: environmental stewardship, sustainable supply chain, employee nurturing, upholding human rights and ethics, economic value creation, and customer centricity. We used these goals to create a framework for sustainability at Neuland.”
Below we explore four of those areas in-depth.
1. Environmental Stewardship
While sustainability emphasizes the need for environmentally responsible products and services, the means to achieve it are often industry- or scale-dependent. A manufacturer, for instance, may have a very different read on sustainable practices than a consultant.
For us, minimizing the impact of manufacturing processes on environmental resources and further preserving them is a key priority. Our aim is to achieve zero landfill waste and become water-positive and carbon neutral by 2030.
Although the pandemic created some hurdles in the development of environmental protection infrastructure and improvement activities, we created a five-fold lower environmental impact in FY 21–22 over FY 20–21. To address greenhouse gas emissions and climate change, we initiated a low carbon pathway plan. Our carbon usage has gone down by 15% after a switch from double- to single-product washing cycles.
We also created a water management strategy focused on lowering water requirements and increasing the use of recycled water in our operations. All of our sites have adopted a “Zero Wastewater Discharge” policy, and we have two-stage high-pressure reverse osmosis systems that recycle 92–94% of our treated wastewater.
 One area of focus for us is improving material efficiency. We’ve seen a 50% reduction in heptane and ethyl acetate use in ezetimibe manufacturing.
One area of focus for us is improving material efficiency. We’ve seen a 50% reduction in heptane and ethyl acetate use in ezetimibe manufacturing.
We are committed to the “wealth from waste” principle. One key target for us has been to achieve “zero land fill” for FY 23. As of August 2022, 100% of our total waste was being sent for recycling and co-processing in the cement industry.
2. Creating Sustainable Supply Chains
Last month, we explored supply chain sustainability and how our supplier assessment matrix includes not only delivery and quality metrics but also environmental and social measurements.
Pharma manufacturing requires key starting materials, intermediates, and specialty materials. The suppliers of these are the focus of Neuland’s sustainable supply chain efforts. Our supply chain team is trained in sustainable procurement practices and plays a key role in our development of domestic supply chains and a diversified vendor base. Our suppliers are required to adhere to a four-part code of conduct including human rights, ethical business practices, regulatory compliance, and safe operation conditions. The result has been geographical de-risking, a shortening of our supply chain, and an increased emphasis on ensuring our supply chain is diverse and inclusive.
3. Safeguarding Employee and Community Wellbeing
Employees are the heart of every company. At Neuland, we employ rigorous audit systems to safeguard employees from occupational injuries. Our team members undergo complete annual health check-ups and medical monitoring programs. To create a culture of sustainable business practices, personnel are trained on ethical compliance practices and behaviors to ensure we are adhering to regulatory policies and maintaining international quality standards.
As part of our broader focus on creating sustainable communities, we work to address community challenges through CSR initiatives targeting education, health, infrastructure development, and water availability.
4. Enabling Customer Interaction & Protecting Their Interests
Customers are a key component of the sustainability matrix. As a result, we seek customer input on specific ESG requirements.
A key aspect of protecting our customers involves the use of digital technologies. Such tools enable closer communication and interaction, improving real-time decision-making. Data-driven analysis allows us to create better, safer, more efficient and more sustainable manufacturing processes.
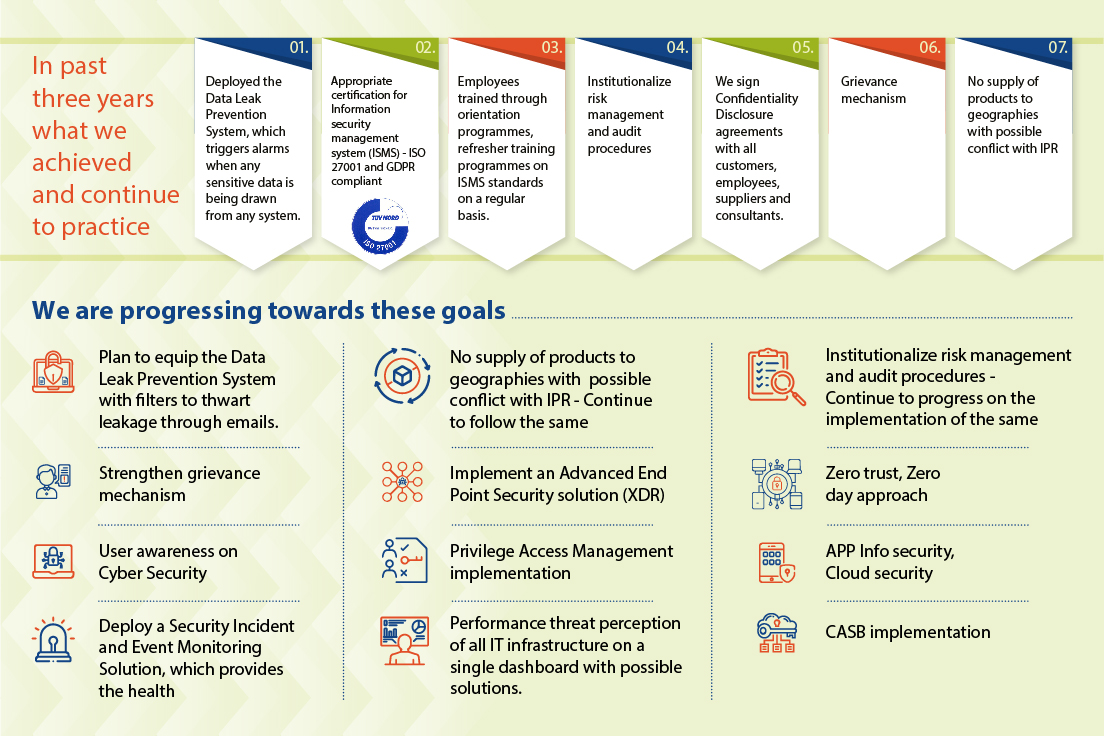
Our growing collective reliance on digital technology to improve our ability to drive value for customers also creates new risks. As an API manufacturer, we understand how crucial our customers’ and partners’ intellectual property rights are, as is the need to maintain strict confidentiality. Our strong digital cybersecurity and data privacy frameworks are designed based on the most relevant and highest-quality standards, and have eliminated any instances of identified leaks, thefts, or losses of customer data as well as security breaches.
Learn more about sustainability at Neuland, or contact us today to discuss your next NCE or generic drug API project with us!








