
 In recent posts on the topic of Quality by Design and drug manufacturing, we referenced (here, here and here) the importance of being able to reduce unanticipated challenges by developing deep process knowledge at the lab scale – which aids in transfer to scale-up.
In recent posts on the topic of Quality by Design and drug manufacturing, we referenced (here, here and here) the importance of being able to reduce unanticipated challenges by developing deep process knowledge at the lab scale – which aids in transfer to scale-up.
We also mentioned that QbD is an effective framework for bringing together a collaborative and inclusive team comprised of both chemists & engineers to ensure a successful API scale-up.
A successful QbD implementation, however, demands more than just a collaborative and inclusive team effort. It also requires three key elements:
- A clear understanding of the target product profile.
- A determination of CQAs to ensure product quality in accordance with regulatory guidelines.
- The design, implementation, and optimization of a manufacturing process using risk assessments, design space (DoE), and process control strategies.
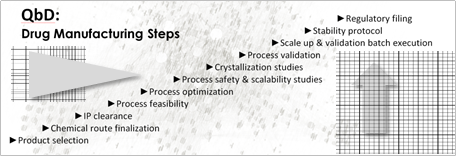
The principal objective underlying a QbD approach to drug manufacturing is developing a comprehensive understanding of the various parameters that can impact the drug candidate and ‘what-if’ scenarios to increase the likelihood of ‘right-at-first-time technology transfer.’
Reducing the Risk of ‘What if…’
While identifying the potentially unexpected is a core element of QbD, so – too – is using the information to reduce the risk of those what-if drug manufacturing scenarios.
Key Criteria for Using the QbD Approach to Minimize Process Uncertainties – the ‘What Ifs’
- Ensure a comprehensive project plan has been developed.
- Implement a periodic review & escalation mechanism for any issues, concerns, etc. relating to process mechanisms (e.g., quality, safety, regulatory, analytical, sourcing).
- Identify clear roles and responsibilities of the cross-functional team.
- Ensure that documentation is complete and accurate – from early process development through technology transfer to manufacturing.
A QbD approach to manufacturing process development should include:
- Identification of the Critical Quality Attributes (CQAs) which impact the drug product.
- Optimization of the manufacturing process based on knowledge of how material attributes and process parameters will affect the drug CQAs.
- Identification of relationships between material attributes, process parameters and CQAs.
- Analysis & assessment of the data to establish product specification ranges.
- Application of Design of Experiments (DOE) to support process development studies, with the objective of reducing the number of experiments needed during development.
Variations in drug substance processes involving chemical conversions can impact the finished drug product. Some of the parameters involved in these processes are controlled, while some are merely noise. To address variations in the process parameters & CQAs, QbD leverages the concept of design space.
The Importance of Communication & Transparency
For a contract manufacturing partner and the client, project challenges can be compounded by long distances and subpar communications. Because more time and effort is invested with a QbD methodology, we’ve found at Neuland that clear communication is crucial. Much can occur during process development and the shift to commercial drug manufacture, and clients tend to want real-time glimpses into the project.
In this way, any concerns can be brought to a client’s notice in a timely manner. Resolutions can be found that tap the requisite expertise needed to overcome the issues – whether in-house or based on Neuland’s experience with similar molecules. In some cases, the project scope is modified in order to adapt to the client’s product development & launch strategies.
In addition to our weekly (or bi-weekly) reviews with the client, we also give our clients full real-time access to the project status through our Critical Chain Project Monitoring (CCPM) management system to maintain full transparency.
This also seems to be a consensus view. From the 2014 Elsevier publication Specification of Drug Substances and Products:
“As to the questions of how much extra work analytical QbD entails, the answer probably ranges from ‘little or nothing’ to ‘a lot,’ depending on how well QbD is built into the method development and validation process. If it comes as an afterthought, it will surely result in extensive extra work. If QbD is built into the process from the beginning, good risk assessment is performed to eliminate low-value studies, and the results of systematic method development are contemporaneously documented. The impact on time and effort should be minimal while increasing method understanding and robustness.”
This is the protocol we follow at Neuland, and we’ve found that the implementation of QbD from the earliest possible stages tends to reduce the possibility of those undesirable ‘what-if’ scenarios.








